Аутоиммунные полигландулярные синдромы
1аиболее часто синдромы полигландулярной недостаточности имеют ауто- [ммунную природу. Аутоиммунные полигландулярные синдромы (АПС) >бусловлены аутоиммунным поражением одновременно двух и более эндо- :ринных желез. В результате, как правило, развивается их недостаточность, 1ередко это сочетается с органоспецифическими неэндокринными заболе- 1аниями аутоиммунного генеза.
На основании клинических и иммуногенетических особенностей выде- гяют АПС-I и АПС-Н (табл. 10.1). Частота поражения отдельных органов ! тканей в рамках одного и того же типа АПС значительно варьирует, что, »ероятно, связано со значительной разницей во времени проявления отдель- шх компонентов этих синдромов.
Таблица 10.1. Аутоиммунные полигландулярные синдромы
При семейных формах проявляется только у сибсов Отсутствие ассоциации с гаплотипом HLA
ГипоПТ, слизисто-кожный кандидоз, хронический активный гепатит, маль- абсорбция Сахарный диабет I типа — 2—5 % Мужчины/женщины — 1,4:1
При семейных формах может проявляться в нескольких поколениях HLA-B8, -Dw3, -DR3, -DR4
Наиболее распространен АПС-Н. Впервые он был описан Шмидтом в 1926 г. как сочетание аддисоновой болезни нетуберкулезной этиологии и аутоиммунного тиреоидита (АИТ). В 1964 г. было отмечено частое сочетание этих заболеваний с инсулинозависимым сахарным диабетом (ИЗСД). Термин “аутоиммунный полигландулярный синдром” был введен Нойфельдом в 1980 г., который определил АПС-П как сочетание первичной хронической надпочечниковой недостаточности с АИТ и/или ИЗСД в отсутствие гипо- паратиреоза и слизисто-кожного кандидоза и тем самым подчеркнул различия АПС-I и АПС-Н. Особенности этих синдромов отражены в табл. 10.2.
В рамках АПС-П могут встречаться и другие заболевания: диффузный токсический зоб (ДТЗ), первичный гипогонадизм, лимфоцитарный гипофи- зит, изолированная недостаточность АКТГ и/или ФСГ и ЛГ; из неэндокринных заболеваний — витилиго, алопеция, пернициозная анемия, миастения, целиакия, герпетиформный дерматит, ювенильный дерматомиозит, изолированный дефицит IgA, аутоиммунная тромбоцитопеническая пурпура, болезнь Паркинсона и др.
Многие из заболеваний, входящие в группу АПС-И, ассоциированы с гаплотипами — HLA-B8, -DR3, -DR4, -DR5. Так, хроническая надпочечниковая недостаточность и ИЗСД в рамках АПС-И ассоциируются с HLA-DR3 и DR4. ДТЗ ассоциирован с HLA-DR3 и -DR5. Изолированный дефицит IgA, ювенильный дерматомиозит и герпетиформный дерматит чаще развиваются у носителей HLA-B8 и -DR3.
В сыворотке крови больных АПС-Н нередко присутствуют органоспецифические аутоантитела к Тг (23,4 %), микросомальной фракции тиреоци- тов (50—58 %), париетальным клеткам желудка (19,8—70 %), островковым клеткам поджелудочной железы (6,2—8 %). Присутствие антител к островковым клеткам на фоне хронической надпочечниковой недостаточности неблагоприятно в прогностическом отношении, поскольку у 8 % таких больных ежегодно развивается ИЗСД. Антитела к ткани яичников встречаются в 3,7—29 % случаев. Высокочувствительными и специфичными маркерами первичной хронической надпочечниковой недостаточности (XHH-I) являются антитела к ферментам стероидогенеза — 21-гидроксилазе (96%), 17а-гидроксилазе (33 %), 20,22-десмолазе (42 %). У 2,3 % больных ИЗСД присутствуют аутоантитела к 21-гидроксилазе, причем их титр значительно выше, чем у больных с изолированной XHH-I. Высокие уровни надпочеч-
эвых аутоантител обнаруживаются в субклинической стадии XHH-I с гедующим снижением их в период выраженных клинических проявле- . Поэтому не исключено, что высокий титр аутоантител к 21-гидрокси- у больных ИЗСД отражает присутствие у них субклинической стадии i-I. Субклиническая фаза XHH-I может продолжаться много лет; клички заболевание проявляется в среднем через 5 лет после обнаружения [тел к клеткам коры надпочечников.
Большинство случаев АПС-Н встречается спорадически, однако неред- [аблюдаются и семейные формы с аутосомно-доминантным типом на- ования при неполной пенетрантности (разное сочетание проявлений gt;11 у разных членов семьи). АПС-П встречается у женщин в 8 раз чаще, у мужчин, и манифестирует обычно в возрасте между 20 и 50 годами, ервал между клиническим дебютом отдельных компонентов синдрома ет превышать 20 лет. У 40—50 % больных с XHH-I рано или поздно ивается другая аутоиммунная эндокринопатия. С другой стороны, у лиц, дающих аутоиммунной патологией щитовидной железы при отсутствии мейном анамнезе АПС-П, риск развития сочетанной эндокринопатии ж.
По данным одного из исследований, АПС-Н был обнаружен у 22 из 44 тентов с XHH-I (16 женщин и 6 мужчин). У 16 из них (73 %) имелись иммунные заболевания щитовидной железы и у 9 (41 %) — ИЗСД. ДТЗ, правило, предшествовал развитию XHH-I, тогда как АИТ во всех слу- развивался после или одновременно с ХНН-1. У 7 из 9 больных ИЗСД шествовал ХНН-1, однако, по другим данным, ИЗСД развивается в нем спустя 7 лет после появления ХНН-1.
В клинической картине АПС-П превалируют проявления ХНН-1. Ги- [игментация при этом может быть выражена слабо, особенно при соче- и с гипотиреозом. Умеренное повышение уровня ТТГ в фазе декомпен- :и ХНН-1 может быть следствием нарушения функции аденогипофиза, гому для диагностики первичного гипотиреоза определение ТТГ необ- мо повторить после компенсации ХНН-1, дополнив его исследованием ня антитиреоидных антител в сыворотке и УЗИ щитовидной железы, очными признаками развития ХНН-1 на фоне ИЗСД являются сниже- цозы инсулина и склонность к гипогликемиям, сочетающиеся с потерей ы тела, диспепсическими расстройствами, гипотензией. ИЗСД после ития гипотиреоза протекает тяжелее. Указаниями на присоединение тиреоза могут служить немотивированная прибавка массы тела, склон- ь к гипогликемиям. ИЗСД в сочетании с ДТЗ отличается лабильным чием и склонностью к кетоацидозу, что в свою очередь способствует итию тиреотоксического криза. Характерной иллюстрацией является ующий случай.
У больной 36 лет, страдающей в течение 10 лет сахарным диабетом, на фоне иьзования относительно небольших доз инсулина появились приступы гипо- 5мии. Последние 1,5 года она стала обращать внимание на потемнение кожных ?вов. Потеря массы тела — 3 кг. Семейный анамнез без особенностей. Обнару- диффузная гиперпигментация кожи, особенно локтевых сгибов, ладонных й, слизистых оболочек губ и десен. АД 120/70 мм рт.ст., ЧСС — 72 в минуту, емия на фоне суточной дозы инсулина 40—44 ЕД колебалась в пределах от 2,7 ,4 ммоль/л. Суточная экскреция свободного кортизола с мочой — 17 нмоль/сут; ;нь АКТГ в 8 ч — 464 пг/мл; содержание тиреоидных гормонов, ТТГ, ПРЛ, ЛГ, , эстрадиола в пределах нормы. При рентгенографии грудной клетки очаговых и инфильтративных изменений не выявлено. Был установлен диагноз XHH-I, и начата терапия кортикостероидами. На фоне терапии преднизолоном (7,5 мг/сут) и кортинеффом (0,05 мг/сут) гипогликемические состояния практически не повторялись.
Через 4,5 мес в связи с очередным ухудшением состояния пациентка снова обратилась в клинику. Гликемия, несмотря на увеличение дозы инсулина до 64 ЕД/сут, — от 11 до 22 ммоль/л. Появились поносы, больная похудела на 4 кг. АД 160/60 мм рт.ст., ЧСС — 120 в минуту. Кожа на ощупь горячая, определяются тремор пальцев вытянутых рук и мелкий тремор закрытых век. При гормональном исследовании: ТТГ — 0,1 мкМЕ/мл, Т4 — 34 пг/мл, Т3 — 9,2 пг/мл. При УЗИ — диффузное увеличение щитовидной железы до 25 мл.
Окончательный диагноз. Аутоиммунный полигландулярный синдром 2-го типа: сахарный диабет I типа средней тяжести в фазе субкомпенсации. Первичная хроническая надпочечниковая недостаточность средней тяжести в фазе компенсации. Диффузный токсический зоб средней тяжести в фазе декомпенсации.
На фоне тиреостатической терапии (мерказолил в стартовой дозе 30 мг/сут) удалось добиться стабилизации состояния. Масса тела увеличилась на 6 кг, гликемия при выписке на фоне привычных доз инсулина — от 5 до 8 ммоль/сут. После радиойодтерапии назначена заместительная терапия L-тироксином (75 мкг/сут).
Сочетание аутоиммунного заболевания щитовидной железы с какой- либо эндокринопатией в отсутствие недостаточности надпочечников некоторые авторы относят к АПС-Ш. Однако большинство все же считают, что такие случаи следует относить к АПС-Н.
АПС-I (APECED — autoimmune polyendocrinopathy-candidiasis-ectoder- mal-dystrphy, MEDAC — multiple endocrine deficiency autoimmune candidiasis, кандидополиэндокринный синдром) — редкое с аутосомно-рецессивным типом наследования или реже — спорадическое заболевание, для которого характерна классическая триада: слизисто-кожный кандидоз, гипопаратиреоз, ХНН-1. Этим заболеваниям могут сопутствовать первичный гипогонадизм и значительно реже — первичный гипотиреоз и ИЗСД. Из неэндокринных заболеваний встречаются пернициозная анемия, витилиго, алопеция, хронический активный гепатит, синдром мальабсорбции, гипоплазия зубной эмали, кератопатия, дистрофия ногтей, эктодермальная дисплазия, аспле- низм, изолированный дефицит IgA, бронхиальная астма, гломерулонефрит.
АПС-I, являясь в целом редкой патологией, с повышенной частотой встречается в финской популяции, среди иранских евреев и сардинийцев, что, вероятно, связано с длительной генетической изоляцией этих народов. Частота новых случаев в Финляндии составляет 1 на 25 000 населения. Этот синдром дебютирует, как правило, в детском возрасте, несколько чаще встречается у мужчин. В описанных семейных случаях заболевание наблюдалось только у сибсов и никогда не встречалась у представителей разных поколений.
В подавляющем большинстве случаев первичным компонентом АПС-1 является слизисто-кожный кандидоз, развивающийся в первые 10 лет жизни, чаще в возрасте около 2 лет. Наблюдается поражение слизистых оболочек полости рта, гениталий, а также кожи, ногтевых валиков, ногтей, реже — ЖКТ и дыхательных путей. На фоне слизисто-кожного кандидоза у 84 % пациентов развивается гипоПТ, чаще всего в первые 10 лет. Кроме характерных судорог мышц конечностей, положительных симптомов Труссо и Хвостека, парестезий, ларингоспазма, у больных наблюдаются судорожные припадки, которые часто расцениваются как проявления эпилепсии. В сред-
нем через 2 года после гипоПТ развивается XHH-I, и у 75 % пациентов она манифестирует в течение 9 лет от начала синдрома. Однако гипокортицизм gt;бычно протекает в латентной форме без выраженной гиперпигментации. Его первым симптомом может оказаться острая недостаточность надпочечников на фоне стрессовой ситуации. Спонтанное смягчение симптомов ипоПТ может служить признаком развития гипокортицизма.
У 10—20 % женщин с АПС-I наблюдается первичный гипогонадизм, появляющийся аменореей. При гормональном исследовании определяются шсокие уровни ЛГ и ФСГ. У мужчин первичный гипогонадизм проявляется iaine всего бесплодием.
По данным одного из исследований, при АПС-I слизисто-кожный сандидоз наблюдался у всех пациентов и в 60 % случаев был первым при- шаком синдрома. ГипоПТ был диагностирован в 79 % случаев, XHH-I — в ^2 %, первичный гипогонадизм у женщин — в 60 % (развивался после 13 тет), у мужчин — в 14 % случаев (развивался после 16 лет). Однако другие 1вторы обнаружили гипопаратиреоз у 22 из 23 больных с АПС-1 (96 %). 8 91 % случаев это заболевание проявлялось до 20-летнего возраста. XHH-I наблюдалась в 22 % случаев и, как правило, развивалась после гипопарати- эеоза. Слизисто-кожный кандидоз наблюдался только у 4 пациентов (17 %), ¦ипогонадизм — у 6 (26 %). Кроме того, у этих больных диагностировали нернициозную анемию, гипотиреоз, алопецию.
Жесткая ассоциация АПС-I с гаплотипами HLA отсутствует. Однако имеется описание слабой ассоциации АПС-I с HLA-A28 и АЗ у финских пациентов. Кроме того, недавно был открыт ген, ответственный за развитие \ПС-1. Он расположен на длинном плече 21-й хромосомы (21q22.3) и солирует, по-видимому, белок AIRE-1 — один из регуляторов транскрип- дии.
Об аутоиммунной природе АПС свидетельствуют лимфоцитарная инфильтрация пораженных желез, присутствие в сыворотке больных органо- шецифических антител и т.п.
У многих больных с АПС-I присутствуют аутоантитела к ферментам надпочечникового стеро идо генеза, а также к антигенам панкреатических
В последнее время охарактеризован и паратиреоидный аутоантиген у Зольных АПС-I. При иммунологическом исследовании крови этих больных эбнаружены также антитела к белкам Candida albicans, которые могут быть использованы как специфичные маркеры кандидоза. Поскольку пациенты : АПС-I редко страдают от распространенного кандидоза, эти антитела
могут играть защитную роль. Специфическими печеночными аутоантигенами при АПС-I могут быть цитохромы Р450 1А2 и Р450 2А6.
Описаны наблюдения 2 семей, у членов которых слизисто-кожный кандидоз сочетался с первичным гипотиреозом. В отличие от АПС-I этот синдром наследуется по аутосомно-доминантному типу. У больных не было обнаружено антител к тиреоидным антигенам и других эндокринопатий. Для своевременного выявления данного синдрома требуется длительное наблюдение за функцией щитовидной железы у всех больных со слизисто-кожным кандидозом.
Изучение возрастно-половой и генетической структуры всех вариантов АПС и, что особенно важно, сопоставление частоты выявления высоких титров органоспецифических антител с различными компонентами этих синдромов позволило выдвинуть единую концепцию формирования аутоиммунных полигландулярных нарушений. Центральное место занимает надпочечниковая недостаточность. При первом варианте АПС болезнь Аддисона манифестирует в молодом возрасте. В этом случае чаще определяются высокие титры антител к антигенам околощитовидных желез (схема 10.1). По возрастно-половой структуре, генетическим маркерам и частоте высоких титров органоспецифических антител сюда же относятся пациенты с изолированным идиопатическим гипоПТ. При втором варианте АПС болезнь Аддисона манифестирует в среднем возрасте. Чаще обнаруживаются высокие титры антител к Тг и другим тиреоидным антигенам. Недостаточность желез иногда не успевает развиться.
Знание закономерностей развития АПС имеет большое практическое значение, поскольку позволяет целенаправленно проводить обследование лиц с заболеваниями, которые могут быть составляющей АПС. Всех больных
ХНН-1 необходимо периодически обследовать на предмет развития у них ИТ и/или первичного гипотиреоза. Нужно также регулярно обследовать гтей, страдающих изолированным идиопатическим гипоПТ, особенно в эчетании с кандидозом, с целью своевременного выявления надпочечни- овой недостаточности. Кроме того, родственникам больных АПС-Н, а акже братьям или сестрам больных АПС-I необходимо 1 раз в несколько ет посещать эндокринолога с целью определения уровня ТТГ, антител к г и микросомальной фракции тиреоцитов, гликемии натощак, уровня онизированного кальция, экскреции свободного кортизола с мочой. Прин- ипы диагностики АПС соответствуют таковым для отдельных составляю- щх их заболеваний.
Лечение АПС заключается в заместительной терапии недостаточности ораженных эндокринных желез. Следует иметь в виду, что при сочетании лпотиреоза с ХНН-1 резкое увеличение дозы L-тироксина может провоци- овать декомпенсацию надпочечниковой недостаточности. Часто приходит- я ограничиваться дозами, не достигающими полных заместительных 1,6 мкг/кг). При сочетании гипоПТ с ХНН-1 следует учитывать, что кортизол витамин D оказывают противоположное влияние на кишечную абсорбцию альция. Таким образом, при дефиците кортизола возрастает риск передо- ировки витамина D. С другой стороны, большие дозы кортикостероидов ри наличии гипоПТ могут спровоцировать выраженную гипокальциемию.
На основании клинических и иммуногенетических особенностей выде- гяют АПС-I и АПС-Н (табл. 10.1). Частота поражения отдельных органов ! тканей в рамках одного и того же типа АПС значительно варьирует, что, »ероятно, связано со значительной разницей во времени проявления отдель- шх компонентов этих синдромов.
Таблица 10.1. Аутоиммунные полигландулярные синдромы
|
АПС-1 |
% |
АПС-Н |
% |
|
ГипоПТ |
79-96 |
Первичный гипокортицизм |
100 |
|
Слизисто-кожный кандидоз |
17-100 |
Первичный гипотиреоз/диффузный токсический зоб |
69-97 |
|
Первичный гипокортицизм |
22-100 |
Сахарный диабет I типа |
35-52 |
|
Первичный гипогонадизм |
26-45 |
Витилиго |
5-50 |
|
Алопеция |
30 |
Первичный гипогонадизм |
3,5-16 |
|
Мальабсорбция |
23 |
Пернициозная анемия |
16 |
|
Пернициозная анемия |
14 |
|
|
|
Хронический активный гепа |
12 |
|
|
|
тит |
|
|
|
|
Первичный гипотиреоз/диф |
10 |
|
|
|
фузный токсический зоб |
|
|
|
|
Витилиго |
4 |
|
|
|
Сахарный диабет I типа |
2-5 |
|
|
| АПС-1 | АПС-Н |
| (пик манифестации — 12 лет) | (пик манифестации — 30 лет) |
При семейных формах проявляется только у сибсов Отсутствие ассоциации с гаплотипом HLA
ГипоПТ, слизисто-кожный кандидоз, хронический активный гепатит, маль- абсорбция Сахарный диабет I типа — 2—5 % Мужчины/женщины — 1,4:1
При семейных формах может проявляться в нескольких поколениях HLA-B8, -Dw3, -DR3, -DR4
Наиболее распространен АПС-Н. Впервые он был описан Шмидтом в 1926 г. как сочетание аддисоновой болезни нетуберкулезной этиологии и аутоиммунного тиреоидита (АИТ). В 1964 г. было отмечено частое сочетание этих заболеваний с инсулинозависимым сахарным диабетом (ИЗСД). Термин “аутоиммунный полигландулярный синдром” был введен Нойфельдом в 1980 г., который определил АПС-П как сочетание первичной хронической надпочечниковой недостаточности с АИТ и/или ИЗСД в отсутствие гипо- паратиреоза и слизисто-кожного кандидоза и тем самым подчеркнул различия АПС-I и АПС-Н. Особенности этих синдромов отражены в табл. 10.2.
В рамках АПС-П могут встречаться и другие заболевания: диффузный токсический зоб (ДТЗ), первичный гипогонадизм, лимфоцитарный гипофи- зит, изолированная недостаточность АКТГ и/или ФСГ и ЛГ; из неэндокринных заболеваний — витилиго, алопеция, пернициозная анемия, миастения, целиакия, герпетиформный дерматит, ювенильный дерматомиозит, изолированный дефицит IgA, аутоиммунная тромбоцитопеническая пурпура, болезнь Паркинсона и др.
Многие из заболеваний, входящие в группу АПС-И, ассоциированы с гаплотипами — HLA-B8, -DR3, -DR4, -DR5. Так, хроническая надпочечниковая недостаточность и ИЗСД в рамках АПС-И ассоциируются с HLA-DR3 и DR4. ДТЗ ассоциирован с HLA-DR3 и -DR5. Изолированный дефицит IgA, ювенильный дерматомиозит и герпетиформный дерматит чаще развиваются у носителей HLA-B8 и -DR3.
В сыворотке крови больных АПС-Н нередко присутствуют органоспецифические аутоантитела к Тг (23,4 %), микросомальной фракции тиреоци- тов (50—58 %), париетальным клеткам желудка (19,8—70 %), островковым клеткам поджелудочной железы (6,2—8 %). Присутствие антител к островковым клеткам на фоне хронической надпочечниковой недостаточности неблагоприятно в прогностическом отношении, поскольку у 8 % таких больных ежегодно развивается ИЗСД. Антитела к ткани яичников встречаются в 3,7—29 % случаев. Высокочувствительными и специфичными маркерами первичной хронической надпочечниковой недостаточности (XHH-I) являются антитела к ферментам стероидогенеза — 21-гидроксилазе (96%), 17а-гидроксилазе (33 %), 20,22-десмолазе (42 %). У 2,3 % больных ИЗСД присутствуют аутоантитела к 21-гидроксилазе, причем их титр значительно выше, чем у больных с изолированной XHH-I. Высокие уровни надпочеч-
эвых аутоантител обнаруживаются в субклинической стадии XHH-I с гедующим снижением их в период выраженных клинических проявле- . Поэтому не исключено, что высокий титр аутоантител к 21-гидрокси- у больных ИЗСД отражает присутствие у них субклинической стадии i-I. Субклиническая фаза XHH-I может продолжаться много лет; клички заболевание проявляется в среднем через 5 лет после обнаружения [тел к клеткам коры надпочечников.
Большинство случаев АПС-Н встречается спорадически, однако неред- [аблюдаются и семейные формы с аутосомно-доминантным типом на- ования при неполной пенетрантности (разное сочетание проявлений gt;11 у разных членов семьи). АПС-П встречается у женщин в 8 раз чаще, у мужчин, и манифестирует обычно в возрасте между 20 и 50 годами, ервал между клиническим дебютом отдельных компонентов синдрома ет превышать 20 лет. У 40—50 % больных с XHH-I рано или поздно ивается другая аутоиммунная эндокринопатия. С другой стороны, у лиц, дающих аутоиммунной патологией щитовидной железы при отсутствии мейном анамнезе АПС-П, риск развития сочетанной эндокринопатии ж.
По данным одного из исследований, АПС-Н был обнаружен у 22 из 44 тентов с XHH-I (16 женщин и 6 мужчин). У 16 из них (73 %) имелись иммунные заболевания щитовидной железы и у 9 (41 %) — ИЗСД. ДТЗ, правило, предшествовал развитию XHH-I, тогда как АИТ во всех слу- развивался после или одновременно с ХНН-1. У 7 из 9 больных ИЗСД шествовал ХНН-1, однако, по другим данным, ИЗСД развивается в нем спустя 7 лет после появления ХНН-1.
В клинической картине АПС-П превалируют проявления ХНН-1. Ги- [игментация при этом может быть выражена слабо, особенно при соче- и с гипотиреозом. Умеренное повышение уровня ТТГ в фазе декомпен- :и ХНН-1 может быть следствием нарушения функции аденогипофиза, гому для диагностики первичного гипотиреоза определение ТТГ необ- мо повторить после компенсации ХНН-1, дополнив его исследованием ня антитиреоидных антител в сыворотке и УЗИ щитовидной железы, очными признаками развития ХНН-1 на фоне ИЗСД являются сниже- цозы инсулина и склонность к гипогликемиям, сочетающиеся с потерей ы тела, диспепсическими расстройствами, гипотензией. ИЗСД после ития гипотиреоза протекает тяжелее. Указаниями на присоединение тиреоза могут служить немотивированная прибавка массы тела, склон- ь к гипогликемиям. ИЗСД в сочетании с ДТЗ отличается лабильным чием и склонностью к кетоацидозу, что в свою очередь способствует итию тиреотоксического криза. Характерной иллюстрацией является ующий случай.
У больной 36 лет, страдающей в течение 10 лет сахарным диабетом, на фоне иьзования относительно небольших доз инсулина появились приступы гипо- 5мии. Последние 1,5 года она стала обращать внимание на потемнение кожных ?вов. Потеря массы тела — 3 кг. Семейный анамнез без особенностей. Обнару- диффузная гиперпигментация кожи, особенно локтевых сгибов, ладонных й, слизистых оболочек губ и десен. АД 120/70 мм рт.ст., ЧСС — 72 в минуту, емия на фоне суточной дозы инсулина 40—44 ЕД колебалась в пределах от 2,7 ,4 ммоль/л. Суточная экскреция свободного кортизола с мочой — 17 нмоль/сут; ;нь АКТГ в 8 ч — 464 пг/мл; содержание тиреоидных гормонов, ТТГ, ПРЛ, ЛГ, , эстрадиола в пределах нормы. При рентгенографии грудной клетки очаговых и инфильтративных изменений не выявлено. Был установлен диагноз XHH-I, и начата терапия кортикостероидами. На фоне терапии преднизолоном (7,5 мг/сут) и кортинеффом (0,05 мг/сут) гипогликемические состояния практически не повторялись.
Через 4,5 мес в связи с очередным ухудшением состояния пациентка снова обратилась в клинику. Гликемия, несмотря на увеличение дозы инсулина до 64 ЕД/сут, — от 11 до 22 ммоль/л. Появились поносы, больная похудела на 4 кг. АД 160/60 мм рт.ст., ЧСС — 120 в минуту. Кожа на ощупь горячая, определяются тремор пальцев вытянутых рук и мелкий тремор закрытых век. При гормональном исследовании: ТТГ — 0,1 мкМЕ/мл, Т4 — 34 пг/мл, Т3 — 9,2 пг/мл. При УЗИ — диффузное увеличение щитовидной железы до 25 мл.
Окончательный диагноз. Аутоиммунный полигландулярный синдром 2-го типа: сахарный диабет I типа средней тяжести в фазе субкомпенсации. Первичная хроническая надпочечниковая недостаточность средней тяжести в фазе компенсации. Диффузный токсический зоб средней тяжести в фазе декомпенсации.
На фоне тиреостатической терапии (мерказолил в стартовой дозе 30 мг/сут) удалось добиться стабилизации состояния. Масса тела увеличилась на 6 кг, гликемия при выписке на фоне привычных доз инсулина — от 5 до 8 ммоль/сут. После радиойодтерапии назначена заместительная терапия L-тироксином (75 мкг/сут).
Сочетание аутоиммунного заболевания щитовидной железы с какой- либо эндокринопатией в отсутствие недостаточности надпочечников некоторые авторы относят к АПС-Ш. Однако большинство все же считают, что такие случаи следует относить к АПС-Н.
АПС-I (APECED — autoimmune polyendocrinopathy-candidiasis-ectoder- mal-dystrphy, MEDAC — multiple endocrine deficiency autoimmune candidiasis, кандидополиэндокринный синдром) — редкое с аутосомно-рецессивным типом наследования или реже — спорадическое заболевание, для которого характерна классическая триада: слизисто-кожный кандидоз, гипопаратиреоз, ХНН-1. Этим заболеваниям могут сопутствовать первичный гипогонадизм и значительно реже — первичный гипотиреоз и ИЗСД. Из неэндокринных заболеваний встречаются пернициозная анемия, витилиго, алопеция, хронический активный гепатит, синдром мальабсорбции, гипоплазия зубной эмали, кератопатия, дистрофия ногтей, эктодермальная дисплазия, аспле- низм, изолированный дефицит IgA, бронхиальная астма, гломерулонефрит.
АПС-I, являясь в целом редкой патологией, с повышенной частотой встречается в финской популяции, среди иранских евреев и сардинийцев, что, вероятно, связано с длительной генетической изоляцией этих народов. Частота новых случаев в Финляндии составляет 1 на 25 000 населения. Этот синдром дебютирует, как правило, в детском возрасте, несколько чаще встречается у мужчин. В описанных семейных случаях заболевание наблюдалось только у сибсов и никогда не встречалась у представителей разных поколений.
В подавляющем большинстве случаев первичным компонентом АПС-1 является слизисто-кожный кандидоз, развивающийся в первые 10 лет жизни, чаще в возрасте около 2 лет. Наблюдается поражение слизистых оболочек полости рта, гениталий, а также кожи, ногтевых валиков, ногтей, реже — ЖКТ и дыхательных путей. На фоне слизисто-кожного кандидоза у 84 % пациентов развивается гипоПТ, чаще всего в первые 10 лет. Кроме характерных судорог мышц конечностей, положительных симптомов Труссо и Хвостека, парестезий, ларингоспазма, у больных наблюдаются судорожные припадки, которые часто расцениваются как проявления эпилепсии. В сред-
нем через 2 года после гипоПТ развивается XHH-I, и у 75 % пациентов она манифестирует в течение 9 лет от начала синдрома. Однако гипокортицизм gt;бычно протекает в латентной форме без выраженной гиперпигментации. Его первым симптомом может оказаться острая недостаточность надпочечников на фоне стрессовой ситуации. Спонтанное смягчение симптомов ипоПТ может служить признаком развития гипокортицизма.
У 10—20 % женщин с АПС-I наблюдается первичный гипогонадизм, появляющийся аменореей. При гормональном исследовании определяются шсокие уровни ЛГ и ФСГ. У мужчин первичный гипогонадизм проявляется iaine всего бесплодием.
По данным одного из исследований, при АПС-I слизисто-кожный сандидоз наблюдался у всех пациентов и в 60 % случаев был первым при- шаком синдрома. ГипоПТ был диагностирован в 79 % случаев, XHH-I — в ^2 %, первичный гипогонадизм у женщин — в 60 % (развивался после 13 тет), у мужчин — в 14 % случаев (развивался после 16 лет). Однако другие 1вторы обнаружили гипопаратиреоз у 22 из 23 больных с АПС-1 (96 %). 8 91 % случаев это заболевание проявлялось до 20-летнего возраста. XHH-I наблюдалась в 22 % случаев и, как правило, развивалась после гипопарати- эеоза. Слизисто-кожный кандидоз наблюдался только у 4 пациентов (17 %), ¦ипогонадизм — у 6 (26 %). Кроме того, у этих больных диагностировали нернициозную анемию, гипотиреоз, алопецию.
Жесткая ассоциация АПС-I с гаплотипами HLA отсутствует. Однако имеется описание слабой ассоциации АПС-I с HLA-A28 и АЗ у финских пациентов. Кроме того, недавно был открыт ген, ответственный за развитие \ПС-1. Он расположен на длинном плече 21-й хромосомы (21q22.3) и солирует, по-видимому, белок AIRE-1 — один из регуляторов транскрип- дии.
Об аутоиммунной природе АПС свидетельствуют лимфоцитарная инфильтрация пораженных желез, присутствие в сыворотке больных органо- шецифических антител и т.п.
У многих больных с АПС-I присутствуют аутоантитела к ферментам надпочечникового стеро идо генеза, а также к антигенам панкреатических
- клеток, в частности, к глутаматдекарбоксилазе и декарбоксилазе аромати- 1еских L-аминокислот. Последние антитела в отличие от антител к глута- натдекарбоксилазе обнаруживаются при АПС-I, но не при ИЗСД. Декар- Зоксилаза ароматических L-аминокислот содержится не только в панкреа- гических (3-клетках, но и в моноаминергических нейронах периферической а центральной нервной системы, печени, почках, коже и энтерохромаффин- -гых клетках кишечника, что, вероятно, может объяснить развитие хрони- 1еского активного гепатита (ХАГ), синдрома мальабсорбции и витилиго у Зольных АПС-I. Действительно, антитела к этому ферменту обнаруживают- ;я у 92 % больных АПС-I, имеющих ХАГ, и лишь у 42 % больных без него; эни выявлялись у 80 % больных АПС-I с витилиго и лишь у 43 % больных Зез этого симптома. Аутоиммунная реакция против гепатоцитов и мелано- дитов кожи может протекать субклинически.
В последнее время охарактеризован и паратиреоидный аутоантиген у Зольных АПС-I. При иммунологическом исследовании крови этих больных эбнаружены также антитела к белкам Candida albicans, которые могут быть использованы как специфичные маркеры кандидоза. Поскольку пациенты : АПС-I редко страдают от распространенного кандидоза, эти антитела
могут играть защитную роль. Специфическими печеночными аутоантигенами при АПС-I могут быть цитохромы Р450 1А2 и Р450 2А6.
Описаны наблюдения 2 семей, у членов которых слизисто-кожный кандидоз сочетался с первичным гипотиреозом. В отличие от АПС-I этот синдром наследуется по аутосомно-доминантному типу. У больных не было обнаружено антител к тиреоидным антигенам и других эндокринопатий. Для своевременного выявления данного синдрома требуется длительное наблюдение за функцией щитовидной железы у всех больных со слизисто-кожным кандидозом.
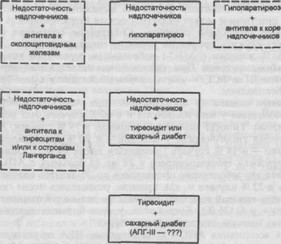
Изучение возрастно-половой и генетической структуры всех вариантов АПС и, что особенно важно, сопоставление частоты выявления высоких титров органоспецифических антител с различными компонентами этих синдромов позволило выдвинуть единую концепцию формирования аутоиммунных полигландулярных нарушений. Центральное место занимает надпочечниковая недостаточность. При первом варианте АПС болезнь Аддисона манифестирует в молодом возрасте. В этом случае чаще определяются высокие титры антител к антигенам околощитовидных желез (схема 10.1). По возрастно-половой структуре, генетическим маркерам и частоте высоких титров органоспецифических антител сюда же относятся пациенты с изолированным идиопатическим гипоПТ. При втором варианте АПС болезнь Аддисона манифестирует в среднем возрасте. Чаще обнаруживаются высокие титры антител к Тг и другим тиреоидным антигенам. Недостаточность желез иногда не успевает развиться.
Знание закономерностей развития АПС имеет большое практическое значение, поскольку позволяет целенаправленно проводить обследование лиц с заболеваниями, которые могут быть составляющей АПС. Всех больных
ХНН-1 необходимо периодически обследовать на предмет развития у них ИТ и/или первичного гипотиреоза. Нужно также регулярно обследовать гтей, страдающих изолированным идиопатическим гипоПТ, особенно в эчетании с кандидозом, с целью своевременного выявления надпочечни- овой недостаточности. Кроме того, родственникам больных АПС-Н, а акже братьям или сестрам больных АПС-I необходимо 1 раз в несколько ет посещать эндокринолога с целью определения уровня ТТГ, антител к г и микросомальной фракции тиреоцитов, гликемии натощак, уровня онизированного кальция, экскреции свободного кортизола с мочой. Прин- ипы диагностики АПС соответствуют таковым для отдельных составляю- щх их заболеваний.
Лечение АПС заключается в заместительной терапии недостаточности ораженных эндокринных желез. Следует иметь в виду, что при сочетании лпотиреоза с ХНН-1 резкое увеличение дозы L-тироксина может провоци- овать декомпенсацию надпочечниковой недостаточности. Часто приходит- я ограничиваться дозами, не достигающими полных заместительных 1,6 мкг/кг). При сочетании гипоПТ с ХНН-1 следует учитывать, что кортизол витамин D оказывают противоположное влияние на кишечную абсорбцию альция. Таким образом, при дефиците кортизола возрастает риск передо- ировки витамина D. С другой стороны, большие дозы кортикостероидов ри наличии гипоПТ могут спровоцировать выраженную гипокальциемию.