
Одномоментньш прорьш плодовнх антигенов через плацен-тарньш барьер или их постепенное неуклонное поступление в течение длительного времени обусловливают различное кли-ническое течение гестоза.
Патологические нарушения при гестозе начинаются с сосу-дисто-тромбоцитарного звена, когда ЦИК фиксируются на клетках эндотелия капилляров, артерий небольшого калибра и на мембранах клеток крови (эритроцитм, тромбоцитм, ней-трофилн и др.). Но прежде всего остановимся на причинах повышенной проницаемости плацентарного барьера для антигенов плода.
Повышение проницаемости маточно-плацентарного барьера происходит чаще всего на фоне снижения маточно-плацентарного кровотока (замедление, остановка, тромбоз в отдельных участках плаценты), при нарушении целостности эпителиального покрова ворсинчатого дерева, который является аналогом эндотелиальной выстилки сосудистой системы.
Иммунные комплексы вызывают изменение гликокалик-са, микроворсинок, цитоплазмы синцитиотрофобласта вплоть до некроза отдельных участков ворсин и целого котиледона. В этих участках и происходит прорыв плацентарного барьера.
Нарушение проницаемости плацентарного барьера для антигенов плода имеет место также при гипоплазии плаценты, вследствие недостаточности инвазии цитотрофобласта в стенки маточно-плацентарных сосудов, при ранней незрелости ворсин, при развитии склероза и редукции просвета сосудов.
Важное значение имеет сочетание уменьшения межворсинчатого пространства с ростом стромальных ворсин, хроническим расстройством маточно-плацентарного кровообращения на фоне снижения синцитиокапиллярных мембран [Милова-нов А. П., 1999].
Таким образом, плацентарная недостаточность различного генеза, сопровождающаяся структурными изменениями плаценты и повышением проницаемости плацентарного барьера, является как бы первым звеном в развитии гестоза. Поэтому профилактика плацентарной недостаточности с 14—16-недельного срока гестации является более эффективным методом предупреждения гестоза, нежели попытки его лечения.
Вернемся к дальнейшим механизмам иммунного повреждения сосудистого эндотелия.
Эндотелиальные клетки сосудистой системы ранее рассматривались как селективный барьер между кровью и тканями. В настоящее время установлено, что эндотелиальные клетки выполняют множество функций (транспортная, метаболическая, биосинтез цитокинов, регуляция гемостаза, поддержание тонуса и проницаемости сосудистой стенки). Основу ан-титромбогенности крови составляют антитромбоцитарная и фибринолитическая активность эндотелия сосудов.
Главная роль эндотелия заключается в сохранении жидкого состояния крови и поддержании гомеостатического баланса "кровь — ткани", включая:
• активный транспорт метаболических субстанций между кровью и тканями;
• образование относительного барьера для макромолекул крови;
• синтез медиаторов, которые регулируют реакцию между сосудистой стенкой и кровью и облегчают гемостатиче-ский гомеостаз;
• фагоцитарную функцию — удаление из крови активных биологических веществ, активированных факторов системы гемостаза, комплемента и др.
Циркулирующие в сосудистом русле тромбоциты не адгези-руют к неповрежденному эндотелию, так как обе структуры имеют одинаково отрицательный электрический заряд.
Если ИК встраивается в сосудистую стенку, отрицательный заряд меняется на положительный. Тромбоциты устремляются к поврежденному участку сосудистой стенки, образуя пристеночные конгломераты и агрегаты.
Неповрежденная сосудистая стенка продуцирует проста-циклин, обладающий сосудорасширяющим и антиагрегант-ным действием. Он же подавляет процесс адгезии и агрегации тромбоцитов. При иммунном воспалении продукция проста-циклина снижается, а в отдельных участках практически прекращается.
Тромбоциты, к мембране которых фиксированы ИК, выбрасывают тромбоксан, который оказывает сильное сосудосуживающее, агрегантное и коагуляционное действие. Фиксация ИК на эндотелии сосудов создает как бы активное поле, на котором происходит пристеночное образование микротромбов. Длительное накопление ИК в крови нарушает систему гемостаза, клеточный метаболизм, повышает перекисное окисление фосфолипидных мембран, снижает антиоксидант-ную защиту.
Организм обедняется фосфолипидами, которые являются основным компонентом всех клеточных мембран. Свидетельством повреждения эндотелия при гестозе служит повышение уровня фибронектина — гликопротеида, входящего в структуру сосудистой стенки.
Иммунное воспаление эндотелия (эндотелиоз) является основной причиной нарушения продукции и соотношения между простациклином и тромбоксаном. Простациклин выделяется также эндотелием сосудов почек, легких, мозга, клетками плодовой части плаценты, капиллярами ворсин, нефроцита-ми. Интенсивность синтеза простациклина связана с антитромбином III, ненасыщенными жирными кислотами, гормонами, эндорфинами и рядом вазоактивных белков.
Продукция простациклина и простагландинов Е повышена при формировании плаценты в первой половине беременности, когда рост плаценты опережает рост плода. Под их влиянием до 20-недельного срока беременности имеет место снижение общего периферического сосудистого сопротивления и некоторое снижение системного артериального давления у беременных на 10—12 мм рт.ст. Оно четко проявляется с 10-не-дельного срока гестации и сохраняется до середины беременности (18—20 нед). Под воздействием простациклина улучшаются реологические свойства крови, увеличивается сердечный выброс, возрастает кровоток в сосудах сердца, почек, плаценты, надпочечников, печени. Все это обеспечивает оптимальные условия для роста плода, а также предупреждает тромбо-тические осложнения в условиях возрастающей активности коагуляционного звена во время беременности.
Тромбоксан и простагландины класса F являются в определенной степени антагонистами простациклина и простаглан-динов Е. Кроме "возбужденного" тромбоцита, тромбоксан выделяется стромой поврежденной сосудистой стенки.
Разнонаправленное действие простациклина и простаглан-дина Е (который является конечным продуктом метаболизма простациклина), а также тромбоксана и простагландина F2a обеспечивает тонко сбалансированный механизм воздействия на сосуды, кровоток, микроциркуляцию и систему гемостаза. Все это обеспечивает необходимые условия для развития плода.
При остром эндотелиозе, который лежит в основе развития гестоза, нарушается динамическое равновесие в продукции простагландинов материнского происхождения (простагландины класса F) и плодово-плацентарной системы (простагландины класса Е). Преобладание простагландинов класса F, увеличение продукции тромбоксана приводят к спазму прекапилляров, артериол, мелких артерий, что сопровождается повышением периферического сосудистого сопротивления и артериальной гипертензией. При гестозе прежде всего повреждаются капилляры, нарушается микроциркуляция, снижается кровоток в почках, печени и плаценте. Наиболее чувствительны к дисбалансу продукции простагландинов F и Е, к снижению содержания простациклина сосуды плаценты, матки, почек, печени, легких и мозга (характерные зоны повреждения при гестозе).
Механизмов снижения продукции простациклинов несколько.
Один из них — недостаточное подавление выработки тромбоксана в плаценте, что имеет место при системной артериальной гипертензии, при затрудненном растяжении матки, при недостаточных децидуальных преобразованиях в сосудах плаценты, при гиперпролактинемии, а также при снижении васкуляризации миометрия.
Используя протеолитический фермент диспазу, японским авторам удалось отделить клетки трофобласта от их базальной мембраны и доказать изолированную продукцию простациклина и тромбоксана различными клетками трофобласта, ворсинками хориона и стенкой артерии. Стенка артерий и артериол плацентн, а также спиральнне артерии субплацентарной зоньт продуцируют как простациклин, так и тромбоксан. При физиологически протекаюхцей беременности количество про-стациклина преобладает над синтезом тромбоксана. При позднем гестозе и гипертензии у беременннх эти соотноше-ния нарушаются в обратной пропорции.
Предполагаемьш механизмом возникновения дисбаланса простагландинов является подавление продукции плацентар-ного простациклина. Это может происходить вследствие воз-действия локального паракринного механизма. Норэпинеф-рин подавляет синтез плацентарного простациклина, но не действует на внработку тромбоксана. Подавление синтеза простациклина происходит опосредованно через метаболитн катехоламинов. Плацента содержит ?2-адренорецепторы связьшаюшие норэпинефрин и эпинефрин. На этот механизм "простациклиновой недостаточности" влияют стресси, изме-нения погодн, нарушение экологии.
На синтез простагландинов в плаценте оказьгаают воздейст-вие гормонн: прогестерон и эстрадиол. Прогестерон подавля-ет синтез тромбоксана и способствует увеличению кровотока в плаценте. Эстрадиол действует противоположно: снижает продукцию простациклина и повншает содержание тромбок-сана. Эти различнме свойства прогестерона и эстрадиола про-являются у женхцин с угрозой прернвания беременности во все сроки (повншенньш тонус матки), когда повьииается про-дукция эстрадиола.
Необходимо подчеркнуть, что иммунньш комплекс, содержа-ший только антиген — антитело, не нарушает структурной цело-стности мембран клеток-мишеней. Нарушение происходит лишь при актавации компонентов комплемента. Несомненно, что в развитии гестоза важную роль играют особенности структурн ан-тигена, имекнцего свойство инициировать острую воспалитель-ную реакцию сосудов. Таким свойством обладают антигенн пло-да, продуцируемьхе в позднефетальном периоде развития.
Если продолжительность циркуляции плодовнх антигенов в организме матери невелика, то даже в случае фиксации ИК на эндотелии сосудов клинические проявления гестоза (отеки, протеинурия, гипертензия) будут транзиторньши и нерезко внраженньши, поскольку ИК разрушаются ретикулоэндоте-лиоцитами печени.
Иммуннме комплексн, которне включают большее содержа-ние плодовьа антигенов, нежеаи антител материнского генеза, а также активизируют сборку комплемента С5 — С9, взаимо-действуют одновременно со множеегвом клеток, содержагцих на своей мембране ?с- и С-рецепторн: эритроцитн, тромбо-цитн, нейтрофильнне гранулоцитн, эозинофилн, базофшш, Т- и В-лимфоцить1, моноцить!, макрофаги. Последствия такого взаимодействия различнм. Разберем некоторме из них.
Схема 1.1 ВНУТРИСОСУДИСТОЕ ПРИСТЕНОЧНОЕ ТРОМБООБРАЗОВАНИЕ
Эритроциты имеют на своей поверхности рецепторы, с помощью которых происходит иммунная адгезия. Эритроциты выводят ИК из циркуляции, транспортируя их в печень, селезенку, где они фагоцитируются. Но при длительном накоплении антигенов плода, их количественном преобладании в структуре комплекса над содержанием антител процесс элиминации нарушается. ИК не просто прикрепляются к поверхности эритроцита, а распластываются на нем, нарушая эластичность и деформабельность мембраны. Эритроцит становится жестким, изменяется его дискоидная форма. Появляются аномальные типы с отростками и острыми выступами (эхиноциты, шизоциты). Они застревают в узких капиллярах, диаметр которых меньше, чем диаметр измененных, нагруженных ИК эритроцитов. Нарушается микроциркуляция, возникает тканевая гипоксия. С увеличением срока длительности гестоза растет кислородная задолженность в окружающих тканях.
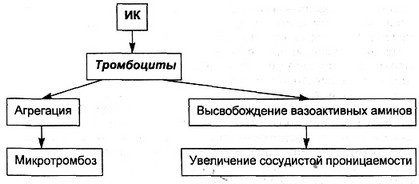
При гестозе прежде всего поражается сосудисто-тромбоци-тарное звено системы гемостаза. Тромбоциты несут на своей поверхности Fc-рецепторы для IgG и для компонентов комплемента. Каждый тромбоцит может фиксировать до 4000 молекул системы комплемента, агрегированного с IgG.
"Возбужденные" тромбоциты высвобождают в кровь тром-боксан, гистамин, серотонин, фактор сосудистой проницаемости, вазоактивные амины, вызывающие сосудистый спазм. Это в свою очередь способствует встраиванию ИК непосредственно в эндотелий и субэндотелиальный слой, а также в мышечный слой артериол и артерий небольшого калибра. Происходит склеивание тромбоцитов внутри сосудистой стенки, формируется процесс внутрисосудистого пристеночного тромбообразования (схема 1.1).
Необходимо подчеркнуть, что потребление тромбоцитов происходит при прогрессировании гестоза, когда развивается ДВС-синдром. Тромбоциты расходуются на образование пристеночных тромбов. Их количество в крови неуклонно снижается. Кроме того, прикрепленные к стенке сосуда тромбоциты увлекаются током крови, ударяются о поврежденные стенки. При этом выделяется не только тромбоксан, но и АДФ. Гиперкоагуляция прогрессирует при постепенном снижении количества тромбоцитов и эритроцитов.
Взаимодействие ИК с нейтрофилами и агрегированным IgG приводит к выделению гранул, из которых выбрасываются сосудосуживающие факторы, высвобождаются протеолитиче-ские ферменты, интерлейкины, вазоактивные пептиды, тром-бопластин, гепарин, анафилактический фактор.
Действие ИК в организме матери исключительно дозозави-симое. Небольшое количество ИК вызывает слабую и непродолжительную воспалительную сосудистую реакцию. Массивное скопление ИК сопровождается тяжелыми и множественными повреждениями жизненно важных органов и систем.
Иммунные комплексы непосредственно участвуют в регуляции иммунного ответа. В малых концентрациях ИК вызывают пролиферацию В-лимфоцитов, в больших — практически подавляют их функциональную активность. ИК способны подавлять активность клеток-киллеров, снижать противоинфек-ционную защиту в материнском организме.
Повреждение эндотелия, повышенный синтез вазоактивных сосудосуживающих веществ (эндотелины, простагландины класса Fa и др.), снижение выделения вазодилататоров и ан-тиагрегантов (простациклин, простагландины класса ly, появление в кровотоке тромбопластических субстанций вызывают первые клинические симптомы гестоза: отеки, гипертен-зию, протеинурию в разном сочетании и слабой степени выраженности.
В тех случаях, когда нового поступления антигена не происходит, процесс формирования ИК не прогрессирует, активированные фагоцитирующие клетки быстро поглощают ИК. Процесс дальнейшего повреждения сосудов, форменных элементов крови и тканей как бы приостанавливается. Следовательно, если прорыв плацентарного барьера уже имел место, но не увеличивается, гестоз принимает легкое и вялотекущее клиническое течение. И напротив, расширение зоны проницаемости в плаценте для антигенов плода приводит к быстрому прогрессиро-ванию осложнения. Однако, если возникновение гестоза имеет место, он далее не останавливается в своем развитии.
Большое значение в патогенезе гестоза имеет активация системы комплемента. Система комплемента состоит из сложного комплекса белков, которые составляют компоненты комплемента С1 — С9, фрагменты их расщепления обозначаются буквами а, Ь, с, (3, §, Г. Например, компонент С5 расхце-пляется на фрагментм С5а и С5Ь.
Синтез компонентов комплемента под воздействием ком-плекса антиген — антитело может происходить медленно или бьгстро. Синтез СЗ происходит со скоростью до 1 мг белка/кг массь! тела в час, что отражается на степени внраженности клинических симптомов гестоза.
Воздействие ИК, содержашего антиген-антитело-активиро-ванньш комплемент, во многом зависит от сочетания компо-нентов белков, вариантов их расшепления и сборки.
Известно, что активация комплемента происходит либо по классическому, либо по альтернативному пуги. Классический пугь активации комплемента заключается в последовательной ак-тивации компоненгов С1, С2, СЗ, С4, вплоть до сборки "мембра-натакуюхцего комплекса" — С5 — С9. Этот комйлекс оказнвает на клетки цитотоксическое и цитолитическое действие, в резуль-тате чего при длительном течении иммунного процесса могут об-разовьшаться дистрофические и некробиотические повреждения органов и систем регуляции, несовместимме с жизнью.
Инициаторами классического пути активации комплемента являются ИК, содержашие чужероднне антигенм, а также фактор XII (Хагемана), тромбопластиновне субстанции, по-павшие в кровь, бактериально-вирусная инфекция, при кото-рой продуцируются 1§О и 1§М (обострение хронической эндо-генной инфекции или острое инфицирование во время бере-менности). Классический путь активации компонентов ком-племента имеет место при длительно текувдем гестозе.
Альтернативньш путь активации комплемента протекает более медленно, но с не менее тяжельши последствиями. Активация компонента по альтернативному пуги происходит под влиянием плазмина, калликреина, трипсина, при воздействии ИК на лей-коцить!, макрофаги, чавде на фоне развившегося ДВС-синдрома.
На уровне образования фрагмента С5Ь, с которого начина-ется образование "мембранатакуюшего комплекса" С5 — С9, оба пути могут состнковьшаться. Комплекс С5 — С9 несет функцию кальциевого канала в тромбоцитах и нейтрофилах и при массивном накоплении этого комплекса в мозговой ткани может возникнуть мгновенная ситуация, когда по этим кана-лам ионь! Са2+ войдут в клетки, внтеснив М§2+, что внзовет отек головного мозга, потерю сознания и обший судорожньш симптом — эклампсию.
Необходимо егце раз подчеркнуть, что эклампсия является са-мьш достоверньш признаком гестоза. Ни одно заболевание (эпилепсия, энцефалопатия и др.) не сопровождается такими характернъши признаками, как приступ эклампсии (отсут-ствие предвестников, мгновенная потеря сознания, асфиксия, тонические и клонические судороги).
Активация фрагаентов комплемента ифает сухцественную роль в развитии гиперкоагуляции через клеточное звено гемо-стаза. В ответ на сборку фрагмента СЗ и С5 моноцитм и эндо-телиальнне клетки секретируют тромбопластин. А образование комплекса С5 — С9 на мембране тромбоцитов вообвде подавля-ет антикоагулянтнне свойства эндотелиальньгх клеток и внзн-вает вмброс тромбопластана и тромбоксана в кровь. Очень бм-стро могут сформироваться пристеночнне тромбн, которне трудно поддежат растворению. Капиллярм закупориваются рас-пластанньши клетками и тромбоцитарньгми массами, снижает-ся кровоток и кровоснабжение органов-мишеней, нарушается микроциркуляция, уменьшаются диффузионнне процессм в различннх участках почек, печени, легких, головном мозге.
Антитромбогеннью свойства эндотелия обусловленн про-стациклиной, которьш постоянно синтезируется и внделяется в кровь. Синтез и секреция простациклина стимулируется брадикинином, тромбином, гистамином, а также калликреи-ном, ангиотензином II, вазопрессином, фрагментом С5а ком-племента. Простациклин обладает внраженньш сосудорасши-ряклцим и антиагрегационньш эффектом. Он расслабляет гладкие мншцн сосудистой стенки и снижает артериальное давление. При повреждении сосудистой стенки ИК (эндоте-лиоз) синтез простациклина снижается. Ангиотензинпревра-1цаю1ций фермент — киназа — превравдает ангиотензин I в мошньш сосудосуживаюхций ангиотензин II, которьш внзн-вает гипертензию. Инактивируется действие брадикинина, плазминогена, что препятствует растворению образовавшихся пристеночннх тромбов.
Актавация системн комплемента приводит к следуюшим биологическим процессам.
^ Миграция лейкоцитов, гранулоцитов, базофилов, моно-цитов, лимфоцитов к поврежденньш участкам эндотелия со-судов и окружаювдих тканей (там, где происходят иммуннне реакции), что проявляется характерньши признаками иммун-ного воспаления.
^ Внсвобождение гистамина из нейтрофшюв, эозинофи-лов, базофилов, которьш повьшает проницаемость сосуди-стой стенки (повншенная гидрофильность тканей, отеки).
^ "Прилипание" циркулируюших иммуннмх комплексов, при которнх клетки крови связьшают компонент комплемента СЗЬ, входятций в ИК (жесткость мембранн эритроцитов, сни-жение деформабельности, повреждение клеточной мембранн тромбоцитов).
^ Компонент комплемента С2 активирует кининовую сис-тему, приводя к развитию диссеминированного внутрисосуди-стого свертьшания крови — ДВС-синдром (при длительно те-кушем гестозе ДВС-синдром формируется почти у 100 % бе-ременннх женвдин).
^ Повреждение мембраны клеток-мишеней под действием "мембранатакующего комплекса" С5 — С9 (проявляется нарушением структуры и функции органов).
Таким образом, активация XII фактора (Хагемана) одновременно включает четыре контактные защитные системы организма (коагуляция, фибринолиз, кининовая система и система комплемента), имеющие между собой самые разные связи и различную активность у каждой пациентки.
Одновременно с формированием ДВС-синдрома активируется фибринолиз, который носит защитный характер, так как растворяет внутрисосудистые микротромбы. За лизис сгустка ответствен фермент плазмин (фибринолизин). Калликреин способен прямо активировать плазминоген, превращая его в плазмин.
Фактор XII играет ведущую роль в индукции субстанций, ответственных за активность иммунной воспалительной реакции сосудов. Его активация усиливает патологические изменения, происходящие при взаимодействии ИК и эндотелиоцитов.
Значение повышенного образования кининов при гестозе заключается в том, что их высокое содержание в крови беременной женщины увеличивает проницаемость первоначально капилляров, далее других кровеносных сосудов и способствует переходу жидкой части крови в межсосудистое пространство (отеки).
Отеки при начавшемся гестозе являются следствием повышения проницаемости капилляров, замедления капиллярного кровотока, увеличения сосудистого сопротивления и повышения капиллярного онкотического давления внутри сосуда, что вызывает выход коллоида в ткани. В более поздние сроки начавшегося и развивающегося гестоза в патологический процесс вовлекаются почечные клубочки, происходят задержка натрия и воды, снижение диуреза, повышение проницаемости почечных клубочков для белка. Клинически это выражается патологической прибавкой массы тела, пастозностью, отеками, которые легко проходят, но также легко рецидивируют.
Через некоторое время к отекам присоединяются транзи-торная гипертензия, следы белка в однократных пробах мочи.
По мере длительности развития гестоза проявляются кли-нико-лабораторные признаки гиперкоагуляции, тромбоцито-пении, ДВС-синдрома.
В заключение этой главы следует подчеркнуть, что патогенез гестоза можно представить в виде айсберга. В надводной, видимой и наименьшей части присутствуют в разном сочетании:
• признаки фетоплацентарной недостаточности (ФПН);
• отеки;
• артериальная гипертензия;
• протеинурия.
В темной глубине человеческого организма (как в подводой части айсберга) произошли и прогрессируют следующие роцессы.
• Повреждения сосудистой эндотелиальной выстилки, обнажение коллагенового слоя, повышение продукции вазокон-стрикторов и агрегантов.
• Снижение деформабельности эритроцитов за счет повышения жесткости и ригидности клеточных мембран, разрушение мембран эритроцитов (гемолиз), тромбоцитов, нейтрофилов. Образование пристеночных тромбов.
• Внутрисосудистая активация свертывания крови и формирование хронического, подострого или острого ДВС-син-дрома.
Повышение проницаемости сосудистых стенок капилляров и выход транссудата в окружающие ткани. Повышение гидрофильности тканей.
• Транзиторный, далее почти постоянный и "жестокий" сосудистый спазм на уровне прекапиллярных сфинктеров, артериол, артерий.
• Гиповолемия, соответствующая недостатку в циркулирующей системе 1200—1500 мл крови.
• Нарушение функции почек.
• Нарушение функции печени.
• Нарушение проницаемости альвеолярных мембран легких — отек легких, инфарктная пневмония, дыхательная недостаточность.
• Аутоиммунная антиорганная агрессия против собственных тканей мозга (эклампсия), плаценты (преждевременная отслойка плаценты), почек и печени (почечно-пече-ночная недостаточность, кортикальный некроз, кровоизлияние в ткань печени).
• Метаболические расстройства: повышение ПОЛ, снижение антиоксидантной защиты.
Гестоз — это иммунокомплексная патология, обусловлен-ая:
• проникновением органоспецифических антигенов плода в кровоток матери;
• образованием иммунных комплексов, в структуру которых входит активированный комплемент;
• нарушением их выведения и разрушения в печени;
• накоплением, циркуляцией и фиксацией ИК на стенках эндотелия сосудов, мембранах форменных элементов крови, в органах с развитой системой микроциркуляции (плацента, печень, легкие, головной мозг);
• формированием острого эндотелиоза, ДВС-синдрома, полиорганной и полисистемной недостаточности.