Системные аномалии развития
Ахондрогенез — один из наиболее тяжелых видов хондродисплазий. Проявляется непропорционально большой головой, запавшей спинкой носа, выраженной микромелией с резким укорочением туловища. Оссификация лобковых и седалищных костей выражена слабо или отсутствует. Выделяют несколько видов:
а) тип Паренти — Фраккаро (син.: ахондрогенез, тип IA; ахондрогенез I типа) — характерны недоношенность, водянка плода. Смерть наступает внутриутробно или вскоре после рождения. У больных короткая шея, бочкообразное туловище и резко укороченные конечности. Голова нормальных размеров, мягкая при пальпации. Рентгенологически выявляются отсутствие оссификации тел позвонков, костей таза, различная степень оссификации костей черепа, короткие ребра, переломы ребер, резкое укорочение и расширение длинных трубчатых костей. Тип наследования - аутосомно-рецессив- ный;
б) тип Лангера — Салдино (син.: ахондрогенез, тип IB; ахондрогенез II типа) - сопровождается водянкой плода и недоношенностью. Голова сильно увеличена, конечности, туловище и шея укорочены. Рентгенологически выявляются недостаточная кальцификация поясничных позвонков и полное отсутствие кальцификации крестца и лобковой кости, нормальная оссификация черепа, сильное укорочение ребер и длинных трубчатых костей, метафизы которых имеют размытые контуры. Укорочение ребер и длинных трубчатых костей выражено слабее, чем при ахондрогенезе I типа. Плод нежизнеспособен. Тип наследования - аутосомно-рецессивный.
Ахондроплазия (син.: Парро — Мари болезнь, хондродисплазия, хондрооистрофия врожденная, хонорооистрофия фетальная, карликовость хонорооистрофическая, карликовость короткоконечностная непропорциональная, микромелия хондродистрофическая фетальная, хондрогенез несовершенный, аплазия диафизарная, ахондроплазия плода, хондродистрофия гипопластическая) - в основе процесса лежит нарушение процесса эн- хондрального остеогенеза, тогда как периостальное окостенение практически не изменено. Хрящ сформирован, но резко гипоплазирован. Клинические признаки: укорочение проксимальных отделов конечностей (ризомелическая микромелия), плюсневых и пястных костей, фаланг пальцев. Кисти широкие и имеют характерную форму, пальцы в виде трезубца, изодактилия. Постоянны микроцефалия, дисплазия лицевого черепа (гипоплазия средней части лица, выступающие лобные бугры, седловидный нос с узкими носовыми ходами, иногда прогнатия). изменения костей таза (укорочение крыльев подвздошных костей, сужение крестцово-подвздошного сочленения, уплощение крыши и неправильные контуры вертлужной впадины). Характерны изменения позвоночника. При относительно нормальной его длине наблюдается симптом сужения расстояния между корнями дужек поясничных позвонков, нарастающий в каудальном направлении. Часто отмечается поясничный кифоз, а позднее может развиться и лордоз. Тип наследования - аутосомно- доминантный.
Вейсмаи — Нетте синдром (Weismann — Netter синдром, син.: dysosteosis Weismann — Netter, dysmorphia diaphysaria tibioflbularis, toxopachyosteosis diaphysaria tibioperonealis) - разновидность наследственного дизостоза: ребенок начинает ходить только в возрасте 2—4 лет; искривление большеберцовой и малоберцовой костей в сагиттальной плоскости: утолщения кортикального вещества диафизов; диспропорциональный низкий или карликовый
рост; деформация V поясничного позвонка, смещение таза; болей и нарушения движений не вызывает. Возможно, аугосомно-рецессивное наследование.
Вролика синдром (Vrolik синдром, син.: osteogenesis imperfecta letalis. osteogenesis imperfecta congenita, fragilitas ossium, osteoporosis fetalis, aplasia periostalis, osteopsatyrosis congenita, morbus Porak — Durante, morbus J'rolik) - наследственная злокачественная форма хрупкости костей: множественные переломы конечностей и ребер уже во время родов; конечности очень короткие и деформированы, с поперечными складками (псевдомикромелия); «каучуковая голова», череп обычной величины, лицо маленькое, незакрывшиеся швы и роднички; часто голубые склеры; изредка плохой слух. Нормальное содержание кальция и фосфора в крови. Карликовый рост, чрезмерно развитые пушковые волосы. Прогноз неблагоприятный — обычно от интеркуррентных заболеваний в течение первых месяцев наступает смерть. Аугосомно-рецессивное наследование.
Гипохондропла нiя сходна с ахондроплазией, но протекает легче. Больные низкого роста, с диспропорционально короткими конечностями за счет проксимальных сегментов. Симптомы проявляются к 3—4-му году жизни. Размеры головы нормальные, иногда отмечается брахицефалия, выступающий лоб. Грудная клетка широкая, плоская, с выступающей грудиной. Кисти и стопы широкие, но пальцы не имеют формы трезубца. Нередко встречаются ограничения движений в тазобедренном и локтевом суставах и варусное искривление голени. Рентгенологически выявляются вогнутые контуры задней поверхности поясничных позвонков, горизонтальная крыша вертлужной впадины, укорочение и утолщение бедренных и плечевых костей, небольшое удлинение малоберцовой кости, «квадратная» форма эпифизов коленных суставов, укорочение локтевой кости в области лучезапястного сустава. Тип наследования предположительно аутосомно-доминантный.
Дизостеосклероз — особая форма остеосклероза. Типичны низкий рост, деформация позвоночника. ломкость костей, нарушение прорезывания постоянных зубов и гипоплазия эмали. В результате склероза костей основания черепа развивается атрофия зрительных и других черепных нервов. При рентгенологическом обследовании выявляются повышенная плотность костей, уплощение и склероз позвонков, расширение метафизов длинных: трубчатых костей с увеличением зоны роста, рассасывание фаланг. Тип наследования аутосомно-рецессивный.
Дисплазия акромезомелическая — характеризуется преимущественным укорочением костей предплечья, кистей и стоп. Наблюдаются дополнительно гипоплазия большеберцовых костей, периферический дизостоз.
Дисплазия диафизарная (син.: Энгельмана болезнь) - истощение, неустойчивая походка, мышечная слабость и быстрая утомляемость, ноющие боли в конечностях и спине, сколиоз и поясничный лордоз, контрактуры суставов, плоскостопие. Рентгенологически обнаруживают симметричное веретенообразное утолщение диафизов длинных трубчатых костей за счет утолщения коркового слоя (диаметр увеличивается в 1,5—2 раза). Участки гиперостоза обычно четко отграничены от метафизов и эпифизов, трабекулы губчатого вещества резко утолщены. Могут поражаться основание черепа и лобные кости, а также ребра, лопатки, ключицы, кости таза, кисти, стопы. Заболевание начинается в возрасте до 30 лет (в большинстве случаев до 10 лет). Тип наследования - аутосомно- доминантный.
Дисплазия кампомелическая (син.: карликовость кампомелическая, искривление костей врожденное) — наиболее типичный признак — кампомелия, проявляющаяся изогнутостью костей голеней, а иногда и бедренных костей. Основной дифференциально-диагнос
тический критерий выявляемое рентгенологически искривление костей. При рентгенографии выявляются также сколиоз, гипоплазия лопаток, короткие и тонкие ключицы, маленькая грудная клетка с узкими ребрами и межреберными промежутками (форма колокола), гипоплазия тел и отростков позвонков, уменьшение крыльев подвздошных костей, относительно широкое тазовое отверстие. Отмечаются укорочение конечностей, эквино- варусное положение стоп, вывих тазобедренных суставов, большой череп с маленькой лицевой частью, плоское лицо, низко расположенные уши, запавшая переносица, гипертелоризм, микрогнатия, глоссоптоз, расщелина неба, умственная отсталость, снижение слуха. Описаны гидроцефалия, аринэнцефалия, лиссэнцефалия, гипоплазия или поликис- тоз почек, гидронефроз, гипоплазия легких, врожденные пороки сердца, гермафродитизм, мужской кариотип у больных с женским фенотипом. Дети, как правило, погибают в периоде новорожденности. Тип наследования аутосомно-рецессивный.
Дисплазия мезомелическая характеризуется укорочением преимущественно костей предплечья и голени. Клинически проявляется ризомелией, гипоплазией нижней челюсти, искривлением конечностей, синостозом костей запястья и предплюсны. Различают несколько типов:
а) дисплазия мезомелическая, тип Лангера (син.: карликовость мезомелическая с гипоплазией локтевой, малоберцовой костей и нижней челюсти) - типична диспропорциональная карликовость за счет укорочения предплечий и голеней. Средний рост — около 130 см. Отмечаются умеренное ограничение разгибания в локтевых суставах, выраженная ульнарная девиация кистей, широкие кисти, брахидактилия. поясничный гиперлордоз, иногда - гипоплазия нижней челюсти. Интеллект в пределах нормы. Рентгенологически выявляют выраженную гипоплазию дистальной части локтевой кости, укорочение, утолщение и искривление лучевой кости. Большеберцовая кость короткая и широкая. Тип наследования предположительно аутосомно-рецессивный;
б) дисплазия мезомелическая. тип Нивергельта проявляется непропорциональной карликовостью с деформацией и укорочением предплечий и голеней. Типичный признак - костные выступы с кожными ямками на боковых поверхностях голеней, иногда на латеральных поверхностях предплечий. Ограничено разгибание в локтевых суставах и суставах пальцев. Кисть отклонена в медиальную сторону. Характерны вальгус- ная деформация локтевых суставов и отведенное эквино-варусное положение стоп. Рост - 135 147 см. Интеллект сохранен. Рентгенологическая картина включает специфичную для синдрома ромбовидную форму большеберцовой и малоберцовой костей, дислокацию и синостоз проксимальной головки лучевой и локтевой костей, синостоз плюсневых костей. Тип наследования — аутосомно-доминантный с различной экспрессивностью;
в) дисплазия мезомелическая, тип Рейнхардта — Пфайффера (син.: гипоплазия локтевой и малоберцовой костей) - рост умеренно снижен, телосложение диспропорциональное. Предплечья укорочены, искривлены в латеральную сторону, ограничены пронация и супинация в локтевых суставах. Отмечается умеренная ульнарная девиация кистей. Голени непропорционально короткие и искривленные, дистальные их части утолщены, на искривленных поверхностях имеются пигментированные кожные ямки. Стопы плоские и нередко отведены в стороны. Рост 150-169 см. Интеллект сохранен. Рентгенологически выявляется укорочение дистальной части локтевой кости; лучевая кость искривлена, ее дистальная часть находит на локтевую; лучезапястный сустав деформирован. Иногда имеется вывих или подвывих головки лучевой кости и дисплазия дистальной суставной поверхности плечевой кости. Проксимальная часть
малоберцовой кости и большеберцовая кость укорочены и искривлены. Тип наследования предположительно аутосомно-доминантный с различной экспрессивностью.
Дисплазия метатропная (син.: карликовость метатропная) - у новорожденных длинная узкая грудная клетка и относительно короткие конечности. Быстро прогрессирует кифо- сколиоз, приводящий со временем к укорочению туловища. Суставы увеличены, движения в них ограничены. Рентгенологически выявляются спондилоэпиметафизарная дисплазия скелета, платиспондилия (в грудном отделе позвонки имеют вид узких «языков»), укорочение ребер, укорочение тела подвздошной кости с «перетяжкой» на границе между телом и крылом, укорочение и расширение шейки бедра, гиперплазия вертельной области, укорочение длинных трубчатых костей с симметричным увеличением метафизов, уплощение и сглаженность контуров эпифизов в области коленных суставов. Дистальные эпифизы костей предплечья, пястных костей и фаланг уменьшены, запаздывает появление ядер окостенения костей запястья. Тип наследования - аутосомно-рецессивный.
Дисплазия метафизарная (син.: Пайла болезнь) - включает высокий рост, диспропорционально длинные нижние конечности, ограничение разгибания в локтевых суставах, валь- гусную деформацию коленных суставов, сколиоз, мышечную слабость, артралгию. склонность к переломам длинных трубчатых костей и иногда аномалии прикуса. Рентгенологически выявляют булавовидное вздутие метафизов длинных и коротких трубчатых костей, медиальных частей ключиц и передних отделов ребер, расширение лобковых и седалищных костей, умеренную платиспондилию, умеренную супраорбитальную гиперплазию. Тип наследования — аутосомно-рецессивный.
Дисплазия парастрелшатическая характеризуется тяжелой асимметричной деформацией скелета. Отмечается резкая задержка роста, платиспондилия, кифосколиоз, асимметричная оссификация тел позвонков, генерализованный остеопороз, гипоплазия и отсутствие оссификации головки бедренной кости, укорочение и искривление длинных трубчатых костей, асимметричная оссификапия и деформация метафизов и эпифизов. Встречается редко.
Дисплазия спондилокостальная (син.: аномалии реберно-вертебральной сегментации, дисплазия споноилоторакальная) — типичен нанизм за счет укорочения туловища, обусловленного множественными аномалиями позвонков и ребер. Череп и конечности не изменены. С возрастом размах рук превышает рост. Нарастает ограничение движений в позвоночнике. Появляются боли в пояснице. Шея укорочена и утолщена, повороты головы и туловища в стороны затруднены. Рентгенологически выявляют нарушение сегментации позвонков, полупозвонки, сагиттальные расщелины позвонков, характерный вид позвонков (бабочковидный позвонок), уменьшение их числа, гипоплазию и слияние ребер. Тип наследования аутосомно-доминантный и аутосомно-рецессивный.
Дисплазия спонд! ьпоэпиметаф! гзарная — характеризуется платиспондилией и дисплазией эпифизов и метафизов трубчатых костей. Постоянны и другие аномалии различных отделов скелета, а также наблюдаются пороки внутренних органов. Встречается редко.
Дисплазия спондаыоэпифизарная в основе процесса лежит дисплазия преимущественно эпифизов костей и позвонков. Различают несколько видов:
а) дисплазия спондилоэпифизарная врожденная — проявляется в двухлетнем возрасте поясничным лордозом и отставанием в росте. Грудная клетка становится бочкоооб- разной. грудина выдается вперед, формируется «утиная» походка. В 50° о случаев выявляется миопия и отслойка сетчатки. Характерны плоское лицо, гипоплазия эмали зубов, гипоплазия мышц (в частности, мышц брюшной стенки), паховые грыжи. Ино
гда отмечаются расщелина неба и косолапость. При нормальной длине конечностей туловище укорочено. Размеры кистей и стоп не изменены. Резко ограничено отведение в тазобедренных суставах. Позже появляются сгибательные контрактуры. Интеллект обычно в норме. Рентгенологически выявляют замедленную оссификацию головок бедренных костей, неправильные волнистые контуры позвонков, с возрастом тела позвонков уплощаются. Отмечаются резкая coxa vara, расширение и разрыхление ростковых зон эпифизов длинных трубчатых костей. В костях кистей и стоп запаздывает появление ядер окостенения. Популяционная частота 0.9:100000. Тип наследования аутосомно-доминантный с различной экспрессивностью;
б) дисплазия спонд! ьпоэпиф! дзарная поздняя — типичен низкий рост за счет укорочения туловища (средний рост взрослых - 120 140 см) при относительно нормальных размерах черепа и конечностей. При рождении длина тела нормальная; в возрасте 5—10 лет рост туловища замедляется, медленно прогрессирует кифосколиоз, появляется «утиная» походка. Шея короткая, грудная клетка широкая. Кисти и стопы обычных размеров. Рано развивающиеся артроз и остеохондроз сопровождаются болями и приводят к ограничению подвижности суставов: иногда отмечается вальгусная деформация голени. Возможна дальнозоркость. Все лабораторные показатели в пределах нормы. Интеллект сохранен. Рентгенологически определяются распространенная платис- пондилия с изменением формы позвонков (вогнутые), кальцификация межпозвоночных дисков, сужение таза, глубокая вертлужная впадина, неравномерность структуры метафизов и дисплазия эпифизов длинных трубчатых костей, признаки остеоспондилита. Популяционная частота — 0,7:100000. Тип наследования — Х-сцепленный рецессивный.
Дисплазия танатофорная (син.: карликовость танатофорная) — длина тела новорожденных обычно 36—46 см. Размеры туловища нормальные, конечности резко укорочены. Пальцы рук короткие, конической формы. Череп относительно большой, с выступающим лбом и запавшей переносицей. Уменьшение размеров грудной клетки приводит к дыхательным расстройствам. У новорожденных отмечаются многочисленные кожные складки, мышечная гипотония и арефлексия. Дети погибают в первые дни жизни, чаще всего от дыхательной недостаточности. Наблюдается следующая рентгенологическая картина: уменьшение высоты тел позвонков, расширение межпозвоночных пространств, уменьшение вертикального и увеличение поперечного размера подвздошной кости, горизонтальное положение нижнего края подвздошной кости, короткие и широкие седалищная и лонные кости, узкая грудная клетка с короткими ребрами, сильно укороченные и относительно широкие длинные трубчатые кости со шпорообразным расширением метафизов, бедренные кости по форме напоминают «телефонную трубку», короткие и широкие кости кистей и стоп. На аутопсии находят сдавление спинного мозга и сужение большого отверстия. Тип наследования аутосомно-доминантный.
Дисплазия фиброзная (син.: Брайцева — Лихтенштейна болезнь, Джеффи — Лихтенштейна болезнь) — замещение компактного слоя кости аваскулярной фиброзной тканью, обусловленное нарушением эмбриогенеза кости на соединительно-тканной стадии. При рентгенологическом исследовании определяются четко отграниченные очаги просветления кости различной величины и формы, соответствующие участкам недифференцированной волокнистой ткани. По локализации различают монооссальную, полиоссальную и регионарную формы, а по характеру изменений в кости очаговую и диффузную формы. Преимущественно поражаются трубчатые кости конечностей, кости черепа. Наблюдаются патологические переломы (рис. 185).
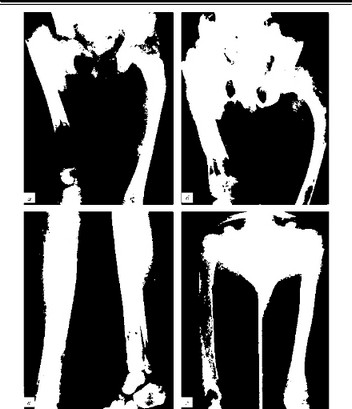
Рис. 185. Полиоссальная форма фиброзной остеодисплазии (Волков М. В., Самойлова JI. И., 1973): а - шейка, межвертельная область и значительная часть диафиза правой бедренной кости утолщены и заняты участками просветления и крапчатыми включениями. Очаги разрежения со структурой «матового стекла» выявляются почти на всем протяжении левой бедренной кости, а также в костях таза;
6 - значительное прогрессирование процесса за 4 года. Увеличилась деформация обоих бедер. Структура костей резко изменена. Бедренные кости расширены, костно-мозговой канал не выражен; в - кости правой голени неравномерно утолщены и деформированы. На всем протяжении структура представлена крупными очагами просветления и участками уплотнения; г - в диафизарных отделах плечевой кости очаги просветления заполняют весь костно-мозговой канал. Кортикальный слой истончен, костно-мозговой канал резко расширен
Дисплазст эпифизарная гемимелическая (син.: тарзомегалия, аклазия тарзоэпифизарная) - односторонняя эпифизарная дисплазия с преимущественной локализацией процесса в таранной кости, костях запястья и предплюсны, а также дистальном (реже в проксимальном) эпифизе большеберцовой и бедренной костей. В основе заболевания лежит чрезмерный рост половины (чаще медиальной) эпифизарного хряща, как правило, одной из указанных костей. Пораженная часть эпифиза утолщается. Иногда возникают вальгусные и варусные деформации суставов. Встречается редко.
Дисплазия эпифизарная множественная в основе лежит дефект центров оссификации эпифизов, в результате чего нарушаются процессы окостенения и образования энхонд- ральной кости, образование же суставного хряща происходит нормально. Различают 2 формы заболевания: легкую, преимущественно позднюю, сопровождающуюся уплощением эпифизов (плоскоэпифизарный тип или тип Роббинга), и тяжелую форму, характеризующуюся малыми по объему эпифизами (тип Фейрбанка). Отмечаются болезненность суставов (в основном тазобедренных), низкий рост, ограничение подвижности в тазобедренных и плечевых суставах, ранний артроз, брахидактилия, нарушение походки, гиперподвижность межфаланговых суставов. Основной диагностический критерий рентгенологические изменения: маленькие, деформированные, уплощенные эпифизы длинных трубчатых костей (чаще всего бедренных и большеберцовых, а также пястных и плюсневых), фрагментация ядер окостенения эпифизов, в единичных случаях - варусная деформация шейки бедра. Метафизы длинных трубчатых костей обычно не изменены, фаланги пальцев укорочены. Позвоночный столб интактный или изменен незначительно. Заболевание проявляется в 2 5 лет. Тип наследования - аутосомно-доминантный с вариабельной экспрессивностью.
Камурати — Энгелъмана синдром (Camurati - Engelman синдром, син.: morbus Camurati - Engelman, hyperosteosis scleroticans systematisata, osteosclerosis hereditaria systematisata, osteopathia hyperosteotica (scleroticans) multiplex infantilis) - склеротический гиперостоз с регредиентной миопатией у детей: прогрессирующая мышечная утомляемость, «утиная походка» у практически здоровых детей. Рентгенологически симметричный генерализованный гиперостоз диафизов длинных трубчатых костей с периостальным склерозом, с нерегулярной нитевидной дегенерацией компактного вещества и сегментированным сужением или расширением костного канала. Спонтанные переломы не наблюдаются. Часто диспропорциональный рост (чрезмерно длинные конечности). Аутосомно-доминант- ное наследование.
Кеффи — Сильвермена синдром (Caffey - Silverman синбром, син.: morbus Coffey' - Silverman, morbus Caffey' - Smith, morbus Roske - de Toni - Caffey - Smith, Toni de - Caffey синдром, Caffey' - de Toni синдром, hyperosteosis corticalis infantilis, poly osteopathia deformans connatalis regressiva, hyperosteogenesis periosteoenchondralis feto-infantilis regressiva) редко встречающийся инфантильный кортикальный гиперостоз: обычно проявляется до 6-месячного возраста. Рентгенологически утолщение компактного и склероз губчатого вещества диафизов длинных и коротких трубчатых костей конечностей, нижней челюсти и ключицы, массивные периостальные наслоения. Умеренно болезненное припухание мягких тканей вокруг пораженной кости; псевдопарезы конечностей. В крови - лейкоцитоз с относительным лимфоцитозом, повышенная СОЭ, анемия, увеличенное содержание фосфатазы. Аутосомно-доминантное наследование.
Клиппеля — Фельдштейна снндром (Klippel - Feldstein синдром, син.: hypertrophia cranialis simplex familiaris) - редкая семейная врожденная скелетная дисплазия: чрезмерно утолщенные кости (особенно ключица и кости черепа); форма черепа оксицефальная или ак-
роцефальная; замедлен рост в длину пальцев рук и ног; подвижность пальцев рук ограничена.
Ламп — Марото синдром (Lamy - Maroteaux синдром, син.: nanismus diastrophicus, дисплазия диастрофическая, синдром диастрофической карликовости) - разновидность наследственного хондродистрофического дизостоза (дистрофический карликовый рост). Наблюдаются диспропорциональный карликовый рост с особенно укороченными проксимальными отделами конечностей, ризомелическая микромелия. Типичны резкое пренатальное отставание в росте, микроцефалия, прогрессирующий сколиоз, кифоз, контрактуры тазобедренных и коленных суставов, выраженная двусторонняя косолапость. В первые месяцы жизни возникает воспаление ушных раковин, после стихания которого у 80° о больных они остаются утолщенными и деформированными. Иногда наблюдается оссификация аурикулярного хряща. Характерна деформация кисти: короткие пальцы с тугоподвижно- стью в межфаланговых суставах II V пальцев и проксимальное расположение большого пальца, изодактилия. В 25° о случаев отмечается расщелина неба. Интеллект сохранен. При рентгенологическом исследовании выявляют сколиоз, укорочение и дугообразную деформацию длинных трубчатых костей, метафизы длинных трубчатых костей расширены, головки бедренных костей деформированы, имеются множественные подвывихи и вывихи в локтевых, тазобедренных и коленных суставах, coxa vara. Постоянные признаки - деформации пястных костей, костей запястья и плюсны, укорочение фаланг пальцев кистей и стоп. Тип наследования - аутосомно-рецессивный.
Лери — Вейля синдром (Leri - Weill синором, син.: Leri синором [III], dysosteosis polytopica enchondralis, дисхондростеоз, карликовость мезомелическая и деформация Маделунга) - разновидность наследственных энхондральных дизостозов, наиболее частая форма мезо- мелической карликовости. Отмечаются диспропорциональный низкий рост, мезомеличе- ское укорочение верхних и нижних конечностей (опущенные руки обычно не достигают тазобедренного сустава), выраженная деформация Маделунга - смещение кисти по отношению к предплечью в ладонном направлении и в локтевую или лучевую сторону. Рентгенологически выявляется изменение дугообразной формы проксимальной суставной линии костей запястья на клиновидную, лучевая кость укорочена, искривлена и скручена по оси, локтевая кость также искривлена, головка ее резко выступает с тыльной стороны. Непостоянные признаки - деформация шейки бедра и плеча, экзостозы проксимальных отделов большой и малой берцовых костей, укорочение и утолщение пястных костей и фаланг, coxa valga, остеоартрит крупных суставов, выраженный лордоз, деформация шейных позвонков. Как осложнение отмечается ограничение подвижности запястья. Тип наследования аутосомно-доминантный.
Лери синдром [I] (Leri синдром [I], син.: Leri - Joanny синдром, morbus Leri, osteosis eburnisans monomelica, melorrheosteosis, osteopathia hyperostotica, ризомономелореостоз) - врожденная наследственная болезнь костей, характеризующаяся резким склерозом, гипе- ростозом и деформацией одной или нескольких длинных трубчатых костей (бедренной, большеберцовой, плечевой). Боль в пораженных суставах, позже ограничение подвижности суставов; часто склеродермия, нередко атрофия и кальциноз мягких тканей; минеральный обмен нормальный. Рентгенологически - продольные полосы кальциноза в пораженных конечностях. Вероятно, аутосомно-рецессивное наследование.
Лери синдром [II] (Leri синором [II], син.: pleonostosis familiaris) - наследственный энхонд- ральный дизостоз: характерный фенотип короткие, толстые пальцы, фиксированные в положении сгибания в I межфаланговом суставе; предплечья в состоянии пронации, плечи ротированы кнутри; супинация и ротация кнаружи невозможны; движения в локтевом
и лучезапястном суставах значительно ограничены; бедра ротированы кнаружи, приведение в тазобедренном суставе невозможно, поэтому невозможно перекрещивание ног; подвижность позвоночного столба ограничена; типичное выражение лица со слегка монголоидными чертами; умственная отсталость, низкий рост. Рентгенологически - диафизы и эпифизы расширены и утолщены. Раннее обызвествление эпифизарного хряща в пястных костях. Андротропизм. Аутосомно-доминантное наследование.
Марото — Ламп синдром [I] (Maroteaitx Lamy синдром [I]) наследственная доброкачественная генерализованная хондродистрофия и дизостоз с дисплазией скелета: диспропорциональный низкий рост с относительно короткими конечностями; краниоцефальная дистрофия - относительно большая голова с выдающимися лобными и затылочными выступами, большой родничок не закрывается вплоть до зрелого возраста: гипоплазия нижней челюсти, аномалии расположения зубов, повышенная склонность к кариесу; нередки множественные спонтанные переломы; аномалии грудной клетки, гипоплазия пальцев, брахидактилия, гипоплазия ногтей. Рентгенологически утолщены и гомогенизированы все кости, особенно утолщено кортикальное вещество длинных костей, расширение швов черепа. Умственное развитие нормальное. Аутосомно-рецессивное наследование.
Марото — Ламп синдром [II] (Maroteaitx - Lamy синдром [II], син.: dysplasia spondylo- epiphysaria tarda) - наследственная спондилоэпифизарная дисплазия: диспропорциональный низкий или карликовый рост (относительное укорочение туловища в связи со сплющенными позвонками) с нормальными по длине конечностями; грудной кифоз, усиленный поясничный лордоз, выпуклая грудина, сужение таза. Рентгенологически генерализованная платиспондилия с деформацией тел позвонков; гипоплазия тазовых костей. Обычно проявляется после 10-летнего возраста. .Андротропизм. Рецессивное, сцепленное с Х-хромосомой наследование.
Мелника — Нндлза синдром (Melnick - Needles синдром, син.: osteodysplasia Melnick - Needles) разновидность наследственной остеодисплазии: недостаточные масса и длина тела при рождении. Сначала развивается диспропорциональный низкий рост, который позже переходит в диспропорциональный нормальный рост. Деформации лица и черепа (легкий гипертелоризм, экзофтальм, оттопыренные ушные раковины, микрогнатия, выпуклый высокий лоб); аномалии расположения зубов и прикуса; запоздалое закрытие родничков; опущенные узкие плечи, тугоподвижность локтевых суставов. Х-образные ноги, вальгусные тазобедренные суставы, гипоплазия больших пальцев рук с выраженным укорочением дистальных фаланг. Деформации тазобедренного сустава и ног уже в раннем детском возрасте вызывают нарушения походки, позже из-за спастической перегрузки развиваются болезненный коксартроз и хромота. Рентгенологически - неравномерно выраженный остеосклероз черепа, преимущественно его основания и пирамиды височной кости; утолщение метафизов и укорочение и S-образное искривление длинных трубчатых костей; неодинаковая толщина коркового вещества костей; дисплазия позвонков, особенно шейных и поясничных; дисплазия лопатки (утолщение и укорочение) и таза (гипоплазия и асимметричность подвздошной, лобковой и седалищной костей); валь- гусное положение тазобедренных суставов. Выраженный гинекотропизм. Предположительно аутосомно-доминантное или доминантное Х-сцепленное наследование.
Метахондроматоз рост низкий; на костях кисти и длинных трубчатых костях отмечаются экзостозы. Остеохондроматозный процесс поражает зоны роста. Со временем возможно обратное развитие патологических изменений в костях. Может наблюдаться ограничение функции пальцев, связанное с экзостозами. Рентгенологически выявляют признаки экзо
стоза и энхондроматоза. В патологический процесс могут вовлекаться тела позвонков. Тип наследования предположительно аутосомно-доминантный.
Нирхофа — Хюбнера синдром (Nierhoff - Hubner синдром) - вариант наследственно! о энхондрального метаэпифизарного дизостоза: микромелия у новорожденного с нормальной длиной тела, приступы выраженных судорог уже в первые дни жизни. Рентгенологически нарушения развития метаэпифизарных частей трубчатых костей, позвонков, ребер, аномалий лица не наблюдается. Летальный исход в первые дни жизни. Аутосомно- доминантное наследование.
Олбрайта синдром (Albright синбром, син.: morbus Albright - McCune - Sternberg, dysplasia polyostotica fibrosa, McCune - Bruch - Albright - Брайцева синдром) - классическую триаду признаков составляют множественная фиброзная дисплазия, пигментация кожи и преждевременное половое созревание. Поражение скелета обычно асимметричное и может быть единственным проявлением заболевания. Рентгенологически в костях обнаруживают неоднородные участки разрежении с псевдокистами. Повреждения имеют сегментарное распределение и могут затрагивать любые части скелета, наиболее часто поражаются кости нижних конечностей, реже верхних конечностей и очень редко - черепа. Возможны деформации и увеличение черепа. У 70° о больных отмечаются асимметричное укорочение нижних конечностей, хромота, переломы, боли при ходьбе, искривление пораженных костей, формирование ложных суставов. Изредка происходит перерождение участков дисплазии в саркому. Почти у 1/3 больных повышен уровень щелочной фосфатазы в крови. Вследствие фиброзной дисплазии черепа могут снижаться зрение и слух. Сразу после рождения на коже обнаруживаются коричневые пигментные пятна с неровными очертаниями, локализующиеся, как правило, в области костной патологии. Распространение пятен одностороннее сегментарное. Преждевременное половое созревание встречается преимущественно у девочек (50° о). Уровень гонадотропинов в моче обычно резко снижен, уровень лютеинизирующего гормона повышен. У некоторых больных повышена экскреция с мочой 17-гидроксикортикостероидов и 17-оксикортико- стероидов. Описаны случаи преждевременного полового созревания у мальчиков. В детстве рост ускорен. Рост взрослых нормальный или несколько ниже среднего. Часто развиваются диффузный или узловатый зоб, умеренный гипертиреоз. Встречается дисплазия паращитовидных желез. Тип наследования неизвестен.
Олье синдром (OUier синдром, син.: morbus OUier, dyschondroplasia Oilier, chondroplasia Oilier, hemichondrodysplasia, энхондроматоз) - наличие в метафизах и диафизах длинных трубчатых костей или в теле плоских костей очагов эмбриональной хрящевой ткани, соответственно которым при рентгенографическом исследовании выявляются различной формы и размеров очаги просветления. В основе лежит замедление и извращение осси- фикации эмбрионального хряща, заключающееся в отсутствии замещения хрящевого скелета костной тканью. Различают по распространенности процесса монооссальную, олигооссальную и полиоссальную формы, а по локализации - 4 формы: акроформу (поражение кистей и стоп), мономелическую (поражение костей одной конечности с прилежащей частью тазового или плечевого пояса), одностороннюю или преимущественно одностороннюю и двустороннюю формы. Наблюдаются укорочение (на 20-30 см) и искривление конечностей, в том числе лучевая и локтевая косорукость, ульнарная девиация запястья, варусное или вальгусное отклонение стопы, утолщение фаланг и ограничение подвижности пальцев. Кроме костных изменений, иногда имеются пигментные пятна или участки депигментации, в единичных случаях у подростков наблюдаются опухоли костей с возможной малигнизацией в зрелом возрасте. Заболевание осложняется переломами по
раженных участков, иногда экзостозами. Рентгенологически - укорочение длинных и коротких трубчатых костей, множественные односторонние энхондромы, видны четко ограниченные очаги овального или веерообразного просветления в метафизах длинных трубчатых костей, занимающие всю толщину кости. При центральном или эксцентричном расположении хондромы кортикальный слой вздут, при боковом расположении опухоли он может разрушаться. В коротких трубчатых костях хрящевые очаги занимают весь диафиз, вызывая его веретенообразное вздутие. Предполагается аутосомно-доминантное наследование. Сочетание с множественными венозными гемангиомами известно под названием синдрома Маффучи.
Остеогенез несовершенный (син.: костеобразование несовершенное) - в основе лежит дефект костеобразования, связанный с недостаточностью мезенхимы. Характеризуется склонностью к переломам длинных трубчатых костей, ребер и ключиц при минимальной травме. Переломы костей черепа, газа и фаланг встречаются крайне редко. Множественные переломы костей приводят к их укорочению и искривлению, образованию ложных суставов; кости голени могут приобретать саблевидную форму. Вследствие деформации конечностей может снижаться рост. Другие скелетные аномалии: кифосколиоз. воронкообразная или килевидная грудная клетка. У больных треугольное лицо, широкий лоб и выступающие виски. Зубы обычно желто-коричневые, опалесцирующие («янтарные»), легко разрушаются. Характерны голубые склеры. На втором десятилетии жизни отосклероз приводит к снижению слуха. Слабость связочного аппарата и мышечная гипотония проявляются разболтанностью суставов и грыжами. Умственное развитие в пределах нормы. Рентгенологически выявляют остеопороз, истончение кортикального слоя, тонкие диафизы с расширенными метафизами, множественные костные мозоли. Тела позвонков имеют двояковогнутую форму («рыбьи позвонки»). Кости черепа истончены, швы расширены, с большим количеством вормиевых костей. Микроскопически отмечаются истончение кортикального слоя трубчатых костей, разрежение и истончение костных балок губчатого вещества. В ряде случаев выявлена дисплазия эпифизов бедренных, большеберцовых и плечевых костей. Фенотипические проявления синдрома вариабельны. В соответствии с современной классификацией выделяют четыре типа несовершенного остеогенеза:
а) I тип - характеризуется относительно доброкачественным течением без выраженных скелетных деформаций;
б) II тип (неонатальный) - проявляется множественными перелом
Источник: Калмин О.В. , «Аномалии развития органов и частей тела человека» 2004