СИЛА И ЭФФЕКТИВНОСТЬ ПРЕПАРАТОВ
Являясь нелинейной, сила эффекта агонистов или антагонистов зависит от количества связанных рецепторов, и, теоретически, максимального эффекта можно достичь, когда все рецепторы связаны.
Наблюдаемые взаимосвязи "доза-эффект" и "концен-трация-эффект" условно представлены полулогарифмически: линейные эффекты нанесены на оси ординат, концентрации - на оси абсцисс. Они имеют типичную сигмоидную форму: эффективность определяется концентрацией (дозой) препарата, требуемой для достижения эффекта. По мере увеличения концентрации агониста молекулы антагониста вытесняются, ингибирование ослабляется, и достигаются те же максимальные эффекты, как и при отсутствии антагониста, но при более высокой концентрации агониста. На графике результирующая кривая агониста в результате влияния антагониста будет сдвинута вправо. Степень этого параллельного сдвига на полулогарифмическом графике зависит от дозы антагониста и активности антагониста. Сдвиг рассчитывают как коэффициент концентраций (доз) агониста, вызывающий одинаковый ответ, как в присутствии, так и в отсутствии антагониста, и называют коэффициентом дозы. Он служит производным концентрации агониста, вызывающей половину максимального эффекта (Е50). Е50 препаратов с аналогичным механизмом действия показывает их относительную силу. Для средств-антагонистов Е50 сравним с IC50 - коэффициентом, представляющим концентрацию, требуемую для ингибирования in vitro на 50%.
На рис. 11.2 продемонстрирована относительная эффективность двух препаратов, ингибирующих один и тот же гипотетический фермент. Препарат А сильнее, чем препарат В, так как для достижения заданного ответа его применяют в меньшей концентрация: кривая "доза-эффект" сдвинута влево по оси Х. Эффект увеличивается до того момента, пока концентрация не достигает максимума, при превышении которого не происходит увеличение эффекта. Это плато определяет эффективность ЛС. Препарат С обладает меньшей силой и эффективностью, чем препараты А и В, а оба этих препарата достигли максимального эффекта и, следовательно, имеют равноценную эффективность, несмотря на разницу в силе. Очевидно, что одновременное назначение препаратов А и В в максимальных дозах не будет более эффективным, чем изолированное применение препарата А или препарата В, что обусловливает тенденцию к использованию средств с различными механизмами действия и молекулярными мишенями в комбинированном лечении ССЗ.
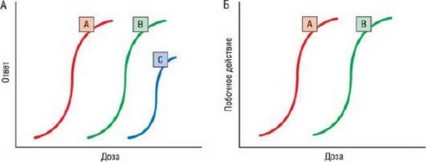
Рис. 11.2. Отношение потенциалов двух ЛС, предположительно ингибирующих один фермент: препарат А имеет большее сродство к ферменту, чем препарат В, так как для достижения эффекта необходима его меньшая концентрация. Препарат С имеет меньшее сродство и менее эффективен, чем препараты А и В.
Умеренные различия в силе между препаратами с одинаковым механизмом действия редко имеют клиническое значение. Во-первых, большинство врачей не учитывают молекулярную массу назначаемых препаратов, без чего невозможно точно сравнить их силу. Например, при сопоставлении по массе амлодипин приблизительно в 6 раз сильнее нифедипина: нифедипин в дозе 60 мг снижает АД в той же степени, что и амлодипин в дозе 10 мг, но при сравнении по молярности амлодипин сильнее почти в 10 раз, что связано с его большей молекулярной массой. Во-вторых, различия в силе редко представляют материальный интерес для врача или пациента до тех пор, пока препараты имеют один и тот же вазодилатирующий эффект: аторвастатин приблизительно в 4 раза сильнее симвастатина (по массе), но большинство дозозависимых эффектов аторвастатина можно воспроизвести с помощью назначения большей дозы симвастатина [5]. Иногда вопрос силы препарата может стать проблемой, если связь его побочных эффектов с концентрацией отличается от терапевтического эффекта. Установлено, что 90% всех нежелательных реакций на препараты служат результатом их основного механизма действия [6]: кривые "доза-ответ" и "доза-нежелательный эффект" будут сдвинуты влево для более сильного препарата (см. рис. 11.2). Есть данные, что это справедливо в отношении статинов: у наиболее сильного препарата этой группы - церивастатина° - лицензия на продажу была отозвана из-за токсичности [7]. С увеличением дозы токсичность аторвастатина становится большей, чем у симвастатина [8].
Нежелательные реакции на препараты не всегда служат следствием их основного механизма действия, а могут быть связаны с их вторичной активностью. В этом случае необходимо подобрать дозу, способную быть эффективной при лечении заболевания и при этом оставаться безопасной. Диапазон между минимальной эффективной дозой препарата и дозой, приводящей к возникновению серьезных побочных эффектов, называют терапевтическим окном. Препараты с узким терапевтическим окном (например, дигоксин) необходимо назначать с осторожностью и контролировать их эффект посредством, например, регулярного измерения концентрации ЛС в крови для предотвращения развития такого побочного эффекта, как аритмия.
ЗАВИСИМОСТЬ "ДОЗА-ЭФФЕКТ"
Клиническая молекулярная фармакология изучает взаимодействие препаратов с их рецепторами. На молекулярном уровне зависимость между концентрацией препарата (на логарифмической шкале) и ответом на него типично сигмоидальная. Клинически ее можно наблюдать при
назначении дозы и получении физиологического ответа (рис. 11.3), хотя зависимость "доза- эффект" in vivo также будет определяться фармакокинетическими параметрами, которые детерминируют концентрацию препарата, фактически достигающую рецептора.
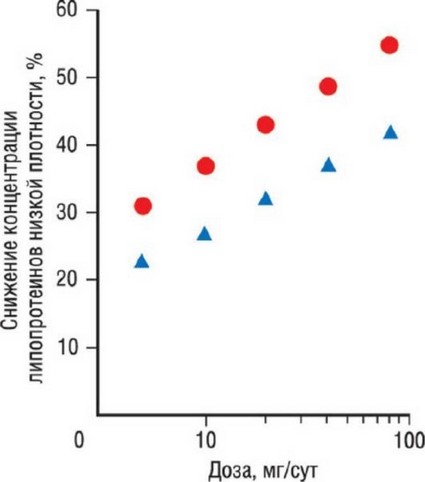
Рис. 11.3. Кривая дозозависимого влияния статинов на концентрацию ХС ЛПНП: аторвастатин обозначен красными кружками, симвастатин - голубыми треугольниками.
В ходе клинического испытания для препарата следует установить зависимость "доза-эффект". Это позволяет определить диапазон пригодных для использования доз и оптимальную дозу, часто определяемую как наименьшую, обладающую максимальной эффективностью. В дальнейшем максимальную эффективность определяют как наибольшее действие препарата, достигнутое на плато зависимости "доза-эффект". При внедрении ЛС в клиническую практику с целью облегчения подбора дозы, необходимой больному, разрешенный диапазон доз следует сужать за счет крутой части кривой. Тем не менее чаще всего препараты вводят в практику в дозах, близких к максимально эффективным. Так, например, обстоит дело с большинством антигипертензивных средств, что не оставляет возможности для увеличения их дозы.
Максимальные эффективные дозы антигипертензивных препаратов близки к вершине дозозависимого диапазона, вне которого дальнейшее увеличение дозы принесет лишь незначительный дополнительный эффект.
Реальные отношения "эффект-доза" в широком диапазоне доз не всегда известны. Установление дозы во II фазе клинических испытаний - одна из самых непростых задач в разработке ЛС. Первоначально каптоприл вводили ежедневно в начальных дозах, превышающих 600 мг, что приводило к существенной гипотензии [9]. В дальнейшем было установлено, что применение
препарата в дозе 150 мг/сут позволило достичь максимального снижения АД при значительно большей безопасности. Тиазидные диуретики много лет использовали в супрамаксимальных гипотензивных дозах до тех пор, пока не было установлено, что пятидесятикратное снижение дозы приводит к тому же эффекту при меньшем числе побочных эффектов [10]. Возможно, для сопоставления эффективности препаратов одного класса в рамках клинического исследования полезным было бы сравнение их зависимости "доза-эффект" (рис. 11.4) [11].
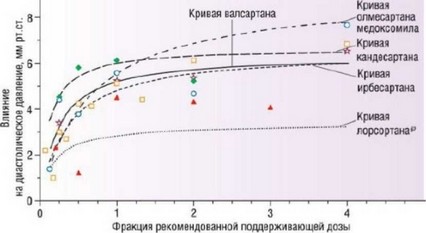
Рис. 11.4. Зависимость "доза-эффект", продемонстрированная на примере БРА [11]: кандесартан обозначен зелеными ромбами, ирбесартан - оранжевыми квадратами, олмесартана медоксомил - синими кружками, валсартан - пурпурными звездами.
ФАРМАКОКИНЕТИКА
Фармакокинетика описывает процессы, происходящие с ЛС, введенными пациенту. В отличие от фармакодинамики, изучающей воздействие препарата на организм, фармакокинетика изучает влияние последнего на ЛС. Область изучения фармакокинетики включает пути и механизмы абсорбции и экскреции, время начала и продолжительность действия препарата, биотрансформацию его молекул в организме и пути экскреции, а также эффекты метаболитов.
АБСОРБЦИЯ
Большинство сердечно-сосудистых препаратов назначают внутрь, что удобно для пациентов, получающих лечение по поводу состояний, приведенных на рис. 11.5. В/в вводят препараты, не абсорбирующиеся из ЖКТ (гепарин натрия) или разрушающиеся в нем (например, белковые средства, такие как тромболитические препараты, несиритидр). В/в введение ЛС рекомендовано в том случае, когда необходимо добиться быстрого начала действия (нитраты, инотропные препараты), важна управляемость дозой препарата в зависимости от эффекта (гепарин натрия для в/в введения у пациентов с высоким риском кровотечений), при недоступности приема внутрь (пациент без сознания) или нарушении функций ЖКТ (например, применение диуретиков при тяжелой СН, когда нет уверенности в их успешной абсорбции в связи с отеком кишечника). Сублингвально применяют препараты, подверженные выраженному и быстрому метаболизму в печени: сублингвальный
прием нитроглицерина позволяет избежать его полной инактивации при первом прохождении через печень. Режим дозирования препаратов определяется их метаболизмом и экскрецией (см. пункты "Метаболизм" и "Экскреция"), а также скоростью абсорбции. Крайне важен интервал дозирования ЛС, поскольку пациенты более привержены лечению, если препарат назначают однократно или два раза в день (указанные режимы имеют схожую комп-лаентность), чем если его применяют чаще [12].
Одно из основных фармакокинетических свойств каждого препарата - биодоступность - описывает долю назначенной дозы препарата, достигающую системного кровотока. По определению, биодоступность препарата, вводимого в/в, достигает 100%, но при другом способе приема (например, внутрь) его биодоступность снижается вследствие неполной абсорбции и метаболизма при первом прохождении через печень.
РАСПРЕДЕЛЕНИЕ
Большинство сердечно-сосудистых препаратов свободно распределяются в сердечно-сосудистой системе и обладают генерализованными эффектами во всех сосудистых бассейнах, где расположены рецепторы и ферменты-мишени. Распределение этих средств за пределами сердечно-сосудистой системы также широкое, хотя и различается для водо- и жирорастворимых веществ. Для жирорастворимых молекул характерен больший объем распределения. Последний, так же известный как видимый объем распределения, - фармакологическая мера распределения препарата в плазме и организме. Объем распределения определяют как объем, необходимый для распределения некоему количеству препарата для достижения определенной концентрации в плазме крови. Почечная недостаточность, в силу задержки жидкости, и печеночная недостаточность, ввиду изменения связывания белков плазмы, могут привести к увеличению объема распределения. Напротив, дегидратация может вызывать снижение объема распределения. Препараты наперстянки и, в большей степени, амиодарон, могут откладываться в жировой ткани в силу крайне высокой жирорастворимости, что приводит к большему объему распределения, необходимости приема нагрузочных доз в начале лечения и длительному периоду выведения при его окончании. Проникновение через гематоэнцефалический барьер жирорастворимого p-адреноблокатора пропранолола может приводить к некоторым побочным эффектам (заметные расстройства сна, ночные кошмары), облегчаемым при переходе на прием водорастворимых препаратов этого же класса (например, атенолола).
МЕТАБОЛИЗМ
Многие сердечно-сосудистые препараты для обретения активности должны подвергнуться метаболизму. К так называемым пролекарствам относят нитраты и большинство иАПФ (например, эналаприл метаболизируется в энаприлат). Метаболизм большинства ЛС приводит к увеличению их водорастворимости, что позволяет препаратам экскретироваться с мочой. Первая фаза реакций приводит к окислению и уменьшению молекулы ЛС, что приводит к потере его активности.
Вторая фаза реакций (конъюгация с глюкуронидом, сульфатами или ацетатами) приводит к обретению водорастворимости и экскреции с мочой. Наиболее интересна первая фаза реакций, протекающая с участием CYP-ферментов - семейством молекул, ответственных за метаболизм большинства антиаритмических препаратов. Генетические вариации CYP-ферментов обнаружены у 10% европейцев, что может приводить к недостаточному метаболизму этих препаратов, их аккумуляции и токсичности [13]. Один из CYP-ферментов, ингибируемый компонентом грейпфрутового сока, ассоциируется с усилением эффекта блокаторов медленных кальциевых каналов [14].
CYP-ферменты обладают распространенным и потенциально важным полиморфизмом. Некоторые из них влияют на метаболизм сердечно-сосудистых препаратов. В числе наиболее важных - вариация фермента CYP2C9, который принимает участие в метаболизме варфарина. Идентифицированы два наиболее часто встречающихся варианта - CYP2C9*2 и CYP2C9*3. Около 20% людей имеют, по крайней мере, один из них. Носительство этих вариантов фермента ассоциируется со сниженной потребностью в варфарине и в 1,5-2 раза более высоким риском возникновения кровотечений. Вопрос о том, будут ли влиять данные о CYP2C9-генотипе на частоту кровотечений в клинической практике, требует проведения проспективных клинических исследований [15].
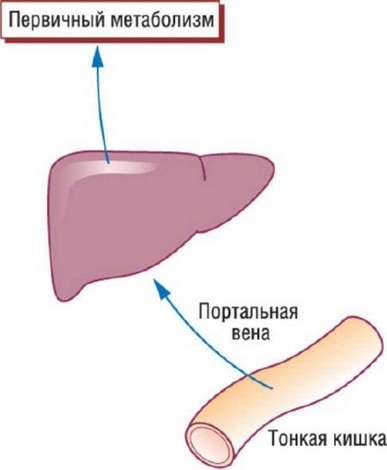
Эффект первого прохождения - феномен метаболизма препарата, посредством которого его концентрация значительно снижается прежде, чем он достигает системного кровотока (рис. 11.6). После приема внутрь ЛС абсорбируется пищеварительной системой и проникает в портальную систему печени. В печени метаболизм множества агентов происходит подчас до такой степени, что лишь небольшая часть активного препарата выходит из органа и достигает циркуляции. Именно поэтому первое прохождение через печень значительно снижает биодоступность. Альтернативой считают в/в введение и сублингвальный прием, позволяющий избежать эффекта первого прохождения через печень для нитратов. Верапамил в значительной степени метаболизируется в печени и уязвим для первого прохождения. p-Адреноблокаторы также подвержены выраженному метаболизму при первом прохождении через печень, поэтому доза атенолола для в/в введения в 10 раз ниже, чем для приема внутрь.
Лекарственный Прием внутрь, Внутривенное _ препарат мг введение, мг
Метопролол 50-100 5-15
Атенолол 50-100 2,5-10
Рис. 11.6. Метаболизм препарата в печени снижает биодоступность в-адреноблокаторов и ацетилсалициловой кислоты при приеме внутрь. При в/в введении препаратов доза должна быть меньше.
Более высокая концентрация ацетилсалициловой кислоты в портальной системе печени, чем в системном кровотоке (в силу метаболизма при первом прохождении), может быть важной в отношении влияния на функцию тромбоцитов. Пока препарат абсорбируется из ЖКТ, большинство циркулирующих тромбоцитов проходят портальную циркуляцию и подвергаются воздействию концентраций ацетилсалициловой кислоты, достаточных для максимального блокирования циклооксигеназы (ЦОГ). В то же время концентрация средства в системном кровотоке, будучи существенно более низкой, может оказывать меньший эффект на эндотелиальную ЦОГ [16].
Наблюдаемые взаимосвязи "доза-эффект" и "концен-трация-эффект" условно представлены полулогарифмически: линейные эффекты нанесены на оси ординат, концентрации - на оси абсцисс. Они имеют типичную сигмоидную форму: эффективность определяется концентрацией (дозой) препарата, требуемой для достижения эффекта. По мере увеличения концентрации агониста молекулы антагониста вытесняются, ингибирование ослабляется, и достигаются те же максимальные эффекты, как и при отсутствии антагониста, но при более высокой концентрации агониста. На графике результирующая кривая агониста в результате влияния антагониста будет сдвинута вправо. Степень этого параллельного сдвига на полулогарифмическом графике зависит от дозы антагониста и активности антагониста. Сдвиг рассчитывают как коэффициент концентраций (доз) агониста, вызывающий одинаковый ответ, как в присутствии, так и в отсутствии антагониста, и называют коэффициентом дозы. Он служит производным концентрации агониста, вызывающей половину максимального эффекта (Е50). Е50 препаратов с аналогичным механизмом действия показывает их относительную силу. Для средств-антагонистов Е50 сравним с IC50 - коэффициентом, представляющим концентрацию, требуемую для ингибирования in vitro на 50%.
На рис. 11.2 продемонстрирована относительная эффективность двух препаратов, ингибирующих один и тот же гипотетический фермент. Препарат А сильнее, чем препарат В, так как для достижения заданного ответа его применяют в меньшей концентрация: кривая "доза-эффект" сдвинута влево по оси Х. Эффект увеличивается до того момента, пока концентрация не достигает максимума, при превышении которого не происходит увеличение эффекта. Это плато определяет эффективность ЛС. Препарат С обладает меньшей силой и эффективностью, чем препараты А и В, а оба этих препарата достигли максимального эффекта и, следовательно, имеют равноценную эффективность, несмотря на разницу в силе. Очевидно, что одновременное назначение препаратов А и В в максимальных дозах не будет более эффективным, чем изолированное применение препарата А или препарата В, что обусловливает тенденцию к использованию средств с различными механизмами действия и молекулярными мишенями в комбинированном лечении ССЗ.
Рис. 11.2. Отношение потенциалов двух ЛС, предположительно ингибирующих один фермент: препарат А имеет большее сродство к ферменту, чем препарат В, так как для достижения эффекта необходима его меньшая концентрация. Препарат С имеет меньшее сродство и менее эффективен, чем препараты А и В.
Умеренные различия в силе между препаратами с одинаковым механизмом действия редко имеют клиническое значение. Во-первых, большинство врачей не учитывают молекулярную массу назначаемых препаратов, без чего невозможно точно сравнить их силу. Например, при сопоставлении по массе амлодипин приблизительно в 6 раз сильнее нифедипина: нифедипин в дозе 60 мг снижает АД в той же степени, что и амлодипин в дозе 10 мг, но при сравнении по молярности амлодипин сильнее почти в 10 раз, что связано с его большей молекулярной массой. Во-вторых, различия в силе редко представляют материальный интерес для врача или пациента до тех пор, пока препараты имеют один и тот же вазодилатирующий эффект: аторвастатин приблизительно в 4 раза сильнее симвастатина (по массе), но большинство дозозависимых эффектов аторвастатина можно воспроизвести с помощью назначения большей дозы симвастатина [5]. Иногда вопрос силы препарата может стать проблемой, если связь его побочных эффектов с концентрацией отличается от терапевтического эффекта. Установлено, что 90% всех нежелательных реакций на препараты служат результатом их основного механизма действия [6]: кривые "доза-ответ" и "доза-нежелательный эффект" будут сдвинуты влево для более сильного препарата (см. рис. 11.2). Есть данные, что это справедливо в отношении статинов: у наиболее сильного препарата этой группы - церивастатина° - лицензия на продажу была отозвана из-за токсичности [7]. С увеличением дозы токсичность аторвастатина становится большей, чем у симвастатина [8].
Нежелательные реакции на препараты не всегда служат следствием их основного механизма действия, а могут быть связаны с их вторичной активностью. В этом случае необходимо подобрать дозу, способную быть эффективной при лечении заболевания и при этом оставаться безопасной. Диапазон между минимальной эффективной дозой препарата и дозой, приводящей к возникновению серьезных побочных эффектов, называют терапевтическим окном. Препараты с узким терапевтическим окном (например, дигоксин) необходимо назначать с осторожностью и контролировать их эффект посредством, например, регулярного измерения концентрации ЛС в крови для предотвращения развития такого побочного эффекта, как аритмия.
ЗАВИСИМОСТЬ "ДОЗА-ЭФФЕКТ"
Клиническая молекулярная фармакология изучает взаимодействие препаратов с их рецепторами. На молекулярном уровне зависимость между концентрацией препарата (на логарифмической шкале) и ответом на него типично сигмоидальная. Клинически ее можно наблюдать при
назначении дозы и получении физиологического ответа (рис. 11.3), хотя зависимость "доза- эффект" in vivo также будет определяться фармакокинетическими параметрами, которые детерминируют концентрацию препарата, фактически достигающую рецептора.
Рис. 11.3. Кривая дозозависимого влияния статинов на концентрацию ХС ЛПНП: аторвастатин обозначен красными кружками, симвастатин - голубыми треугольниками.
В ходе клинического испытания для препарата следует установить зависимость "доза-эффект". Это позволяет определить диапазон пригодных для использования доз и оптимальную дозу, часто определяемую как наименьшую, обладающую максимальной эффективностью. В дальнейшем максимальную эффективность определяют как наибольшее действие препарата, достигнутое на плато зависимости "доза-эффект". При внедрении ЛС в клиническую практику с целью облегчения подбора дозы, необходимой больному, разрешенный диапазон доз следует сужать за счет крутой части кривой. Тем не менее чаще всего препараты вводят в практику в дозах, близких к максимально эффективным. Так, например, обстоит дело с большинством антигипертензивных средств, что не оставляет возможности для увеличения их дозы.
Максимальные эффективные дозы антигипертензивных препаратов близки к вершине дозозависимого диапазона, вне которого дальнейшее увеличение дозы принесет лишь незначительный дополнительный эффект.
Реальные отношения "эффект-доза" в широком диапазоне доз не всегда известны. Установление дозы во II фазе клинических испытаний - одна из самых непростых задач в разработке ЛС. Первоначально каптоприл вводили ежедневно в начальных дозах, превышающих 600 мг, что приводило к существенной гипотензии [9]. В дальнейшем было установлено, что применение
препарата в дозе 150 мг/сут позволило достичь максимального снижения АД при значительно большей безопасности. Тиазидные диуретики много лет использовали в супрамаксимальных гипотензивных дозах до тех пор, пока не было установлено, что пятидесятикратное снижение дозы приводит к тому же эффекту при меньшем числе побочных эффектов [10]. Возможно, для сопоставления эффективности препаратов одного класса в рамках клинического исследования полезным было бы сравнение их зависимости "доза-эффект" (рис. 11.4) [11].
Рис. 11.4. Зависимость "доза-эффект", продемонстрированная на примере БРА [11]: кандесартан обозначен зелеными ромбами, ирбесартан - оранжевыми квадратами, олмесартана медоксомил - синими кружками, валсартан - пурпурными звездами.
ФАРМАКОКИНЕТИКА
Фармакокинетика описывает процессы, происходящие с ЛС, введенными пациенту. В отличие от фармакодинамики, изучающей воздействие препарата на организм, фармакокинетика изучает влияние последнего на ЛС. Область изучения фармакокинетики включает пути и механизмы абсорбции и экскреции, время начала и продолжительность действия препарата, биотрансформацию его молекул в организме и пути экскреции, а также эффекты метаболитов.
АБСОРБЦИЯ
Большинство сердечно-сосудистых препаратов назначают внутрь, что удобно для пациентов, получающих лечение по поводу состояний, приведенных на рис. 11.5. В/в вводят препараты, не абсорбирующиеся из ЖКТ (гепарин натрия) или разрушающиеся в нем (например, белковые средства, такие как тромболитические препараты, несиритидр). В/в введение ЛС рекомендовано в том случае, когда необходимо добиться быстрого начала действия (нитраты, инотропные препараты), важна управляемость дозой препарата в зависимости от эффекта (гепарин натрия для в/в введения у пациентов с высоким риском кровотечений), при недоступности приема внутрь (пациент без сознания) или нарушении функций ЖКТ (например, применение диуретиков при тяжелой СН, когда нет уверенности в их успешной абсорбции в связи с отеком кишечника). Сублингвально применяют препараты, подверженные выраженному и быстрому метаболизму в печени: сублингвальный
прием нитроглицерина позволяет избежать его полной инактивации при первом прохождении через печень. Режим дозирования препаратов определяется их метаболизмом и экскрецией (см. пункты "Метаболизм" и "Экскреция"), а также скоростью абсорбции. Крайне важен интервал дозирования ЛС, поскольку пациенты более привержены лечению, если препарат назначают однократно или два раза в день (указанные режимы имеют схожую комп-лаентность), чем если его применяют чаще [12].
Одно из основных фармакокинетических свойств каждого препарата - биодоступность - описывает долю назначенной дозы препарата, достигающую системного кровотока. По определению, биодоступность препарата, вводимого в/в, достигает 100%, но при другом способе приема (например, внутрь) его биодоступность снижается вследствие неполной абсорбции и метаболизма при первом прохождении через печень.
РАСПРЕДЕЛЕНИЕ
Большинство сердечно-сосудистых препаратов свободно распределяются в сердечно-сосудистой системе и обладают генерализованными эффектами во всех сосудистых бассейнах, где расположены рецепторы и ферменты-мишени. Распределение этих средств за пределами сердечно-сосудистой системы также широкое, хотя и различается для водо- и жирорастворимых веществ. Для жирорастворимых молекул характерен больший объем распределения. Последний, так же известный как видимый объем распределения, - фармакологическая мера распределения препарата в плазме и организме. Объем распределения определяют как объем, необходимый для распределения некоему количеству препарата для достижения определенной концентрации в плазме крови. Почечная недостаточность, в силу задержки жидкости, и печеночная недостаточность, ввиду изменения связывания белков плазмы, могут привести к увеличению объема распределения. Напротив, дегидратация может вызывать снижение объема распределения. Препараты наперстянки и, в большей степени, амиодарон, могут откладываться в жировой ткани в силу крайне высокой жирорастворимости, что приводит к большему объему распределения, необходимости приема нагрузочных доз в начале лечения и длительному периоду выведения при его окончании. Проникновение через гематоэнцефалический барьер жирорастворимого p-адреноблокатора пропранолола может приводить к некоторым побочным эффектам (заметные расстройства сна, ночные кошмары), облегчаемым при переходе на прием водорастворимых препаратов этого же класса (например, атенолола).
МЕТАБОЛИЗМ
Многие сердечно-сосудистые препараты для обретения активности должны подвергнуться метаболизму. К так называемым пролекарствам относят нитраты и большинство иАПФ (например, эналаприл метаболизируется в энаприлат). Метаболизм большинства ЛС приводит к увеличению их водорастворимости, что позволяет препаратам экскретироваться с мочой. Первая фаза реакций приводит к окислению и уменьшению молекулы ЛС, что приводит к потере его активности.
Вторая фаза реакций (конъюгация с глюкуронидом, сульфатами или ацетатами) приводит к обретению водорастворимости и экскреции с мочой. Наиболее интересна первая фаза реакций, протекающая с участием CYP-ферментов - семейством молекул, ответственных за метаболизм большинства антиаритмических препаратов. Генетические вариации CYP-ферментов обнаружены у 10% европейцев, что может приводить к недостаточному метаболизму этих препаратов, их аккумуляции и токсичности [13]. Один из CYP-ферментов, ингибируемый компонентом грейпфрутового сока, ассоциируется с усилением эффекта блокаторов медленных кальциевых каналов [14].
CYP-ферменты обладают распространенным и потенциально важным полиморфизмом. Некоторые из них влияют на метаболизм сердечно-сосудистых препаратов. В числе наиболее важных - вариация фермента CYP2C9, который принимает участие в метаболизме варфарина. Идентифицированы два наиболее часто встречающихся варианта - CYP2C9*2 и CYP2C9*3. Около 20% людей имеют, по крайней мере, один из них. Носительство этих вариантов фермента ассоциируется со сниженной потребностью в варфарине и в 1,5-2 раза более высоким риском возникновения кровотечений. Вопрос о том, будут ли влиять данные о CYP2C9-генотипе на частоту кровотечений в клинической практике, требует проведения проспективных клинических исследований [15].
Эффект первого прохождения - феномен метаболизма препарата, посредством которого его концентрация значительно снижается прежде, чем он достигает системного кровотока (рис. 11.6). После приема внутрь ЛС абсорбируется пищеварительной системой и проникает в портальную систему печени. В печени метаболизм множества агентов происходит подчас до такой степени, что лишь небольшая часть активного препарата выходит из органа и достигает циркуляции. Именно поэтому первое прохождение через печень значительно снижает биодоступность. Альтернативой считают в/в введение и сублингвальный прием, позволяющий избежать эффекта первого прохождения через печень для нитратов. Верапамил в значительной степени метаболизируется в печени и уязвим для первого прохождения. p-Адреноблокаторы также подвержены выраженному метаболизму при первом прохождении через печень, поэтому доза атенолола для в/в введения в 10 раз ниже, чем для приема внутрь.
Лекарственный Прием внутрь, Внутривенное _ препарат мг введение, мг
Метопролол 50-100 5-15
Атенолол 50-100 2,5-10
Рис. 11.6. Метаболизм препарата в печени снижает биодоступность в-адреноблокаторов и ацетилсалициловой кислоты при приеме внутрь. При в/в введении препаратов доза должна быть меньше.
Более высокая концентрация ацетилсалициловой кислоты в портальной системе печени, чем в системном кровотоке (в силу метаболизма при первом прохождении), может быть важной в отношении влияния на функцию тромбоцитов. Пока препарат абсорбируется из ЖКТ, большинство циркулирующих тромбоцитов проходят портальную циркуляцию и подвергаются воздействию концентраций ацетилсалициловой кислоты, достаточных для максимального блокирования циклооксигеназы (ЦОГ). В то же время концентрация средства в системном кровотоке, будучи существенно более низкой, может оказывать меньший эффект на эндотелиальную ЦОГ [16].
Источник: Кэмм А. Джон, Люшер Томас Ф., Серруис П.В., «Болезни сердца и сосудов.Часть 3 (Главы 11-15)» 2011
А так же в разделе « СИЛА И ЭФФЕКТИВНОСТЬ ПРЕПАРАТОВ »
- РЕЗЮМЕ
- ОСНОВЫ КЛИНИЧЕСКОЙ ФАРМАКОЛОГИИ
- ЭКСКРЕЦИЯ
- ПЛЕОТРОПНЫЕ ЭФФЕКТЫ ПРЕПАРАТОВ
- ПОБОЧНЫЕ ЭФФЕКТЫ СЕРДЕЧНО-СОСУДИСТЫХ ПРЕПАРАТОВ
- ВЛИЯНИЕ НЕКАРДИОЛОГИЧЕСКИХ ПРЕПАРАТОВ НА СЕРДЕЧНО-СОСУДИСТУЮ СИСТЕМУ
- КАРДИОТОКСИЧЕСКИЕ ПРЕПАРАТЫ
- ФАРМАКОГЕНОМИКА, ФАРМАКОГЕНЕТИКА И СЕРДЕЧНО-СОСУДИСТЫЕ ПРЕПАРАТЫ
- ПРЕПАРАТЫ, ВЛИЯЮЩИЕ НА РЕНИН-АНГИОТЕНЗИН-АЛЬДОСТЕРОНОВУЮ СИСТЕМУ
- ПОЛЬЗА И РИСК НАЗНАЧЕНИЯ СЕРДЕЧНО-СОСУДИСТЫХ ПРЕПАРАТОВ ОТДЕЛЬНЫМ БОЛЬНЫМ