Талассемии
Талассемии - гетерогенная группа наследственно обусловленных заболеваний, в основе которых лежит нарушение синтеза одной из полипептид- ных цепей глобина, что приводит к увеличению продукции других цепей и развитию дисбаланса между ними. Талассемии относят к количественным гемоглобинопатиям, так как структура цепей гемоглобина не изменена. Различают а-талассемию, когда нарушается синтез а-непей, и р-талассемию при блокаде синтеза P-цепей глобина. Описаны также случаи у-, б- и рб- талассемии с нарушением синтеза соответствующих цепей глобина. Чаще встречаются Р-талассемии. Цепи, синтезируемые в избьггочном количестве, накапливаются и откладываются в эритрокариоцитах костного мозга и эритроцитах периферической крови, вызывая повреждение клеточной мембраны и преждевременную гибель клеток. Эритрокариоциты гибнут в селезенке, костном мозге. Анемия сопровождается небольшим повышением ретикуло- цитов. Дисбаланс синтеза глобиновых цепей вызывает развитие неэффективного эритропоэза, внутриклеточный гемолиз эритроцитов периферической крови, гиперспленизм и развитие гипохромной анемии различной степени тяжести.
Р-талассемия. Р-талассемия - гетерогенное заболевание. В настоящее время известно более 100 мутаций, вызывающих р-талассемию. Обычно дефект состоит в образовании неполноценной мРНК Р-глобина. Разнообразие молекулярных дефектов приводит к тому, что так называемая гомозиготная Р-талассемия нередко представляет двойное гетерозиготное состояние по разным дефектам синтеза Р-глобина. Различают Р°-талассемию, когда у гомозигот полностью отсутствует синтез p-цепей глобина, и р+-талассемию, при частично сохраненном синтезе p-цепей. Среди р+-талассемий выделяют две основные формы: тяжелую средиземноморскую форму, при которой синтезируется около 10% нормальной цепи (большая талассемия, анемия Кули), и более легкую негритянскую форму, когда сохраняется около 50% синтеза нормальной P-цепи. В г руппу Р-талассемий относят также 5р-талассемию и Hb Lepore. В результате этого имеются существенные расхождения в клинической картине различных форм талассемии, однако для всех р-талассемий общим является внутриклеточный гемолиз эритроцитов, неэффективный эритропоэз в костном мозге и спленомегалия.
Большая талассемия (анемия Кули, thalassemia major). Считается гомозиготной формой талассемии, хотя во многих случаях заболевание является двойным гетерозиготным состоянием по различным формам р-талассемии. Клинически заболевание проявляется к концу 1-2-го года жизни ребенка спленомегалией, желтухой, бледностью кожных покровов, изменением костей - квадратный череп, уплощенная переносица, выступающие скулы, сужение глазных щелей. Дети физически плохо развиты.
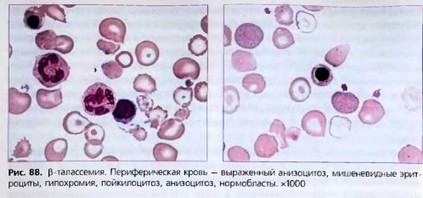
В костном мозге наблюдается гиперплазия красного ростка, выявляется значительное количество сидеробластов. В крови - гипохромная микроцитарная анемия (снижены MCV, МСН, МСНС), резкий анизоцитоз, встречаются эритроциты с базофильной пунктацией, эритрокариоциты, пойкилоцитоз, мишеневидные эритроциты, шизоциты (рис. 88). Даже при тяжелой анемии количество ретикулоцитов не бывает высоким, так как в костном мозге выражен неэффективный эритропоэз. Отмечается повышение осмотической резистентности эритроцитов. Характерна лейкопения с относительным лим- фоцитозом, в период гемолитического криза - нейтрофильный лейкоцитоз со сдвигом влево. В период криза может наблюдаться левый сдвиг.
В сыворотке крови имеет место гипербилирубинемия за счет неконъ- югированного билирубина, повышено содержание сывороточного железа. Избыточное отложение железа приводит к сидерозу органов. Характерным
признаком большой талассемии является выраженное увеличение концентрации фетального гемоглобина. Количество НЬА варьирует в зависимости от типа талассемии. У гомозигот с Р°-талассемией НЬА практически отсутствует. При р+-талассемии (средиземноморский тип) НЬА варьирует от 10 до 25%, при Р+-талассемии негритянского типа содержание НЬА значительно выше. Однако тяжесть заболевания не всегда коррелирует с количеством фетального гемоглобина. Содержание НЬА2 может быть различным, чаще повышенным, но отношение НЬА2/ НЬА всегда меньше, чем 1:40. Диагноз подтверждается электрофорезом гемоглобина (уровень HbF - до 70%).
Малая талассемия (thalassemia minor) является гетерозиготной формой Р-талассемии. Клинически малая талассемия характеризуется менее выраженными симптомами, чем большая талассемия, может протекать практически бессимптомно.


В костном мозге - гиперплазия эритроидного ростка, количество си- деробластов повышено или нормальное. В крови наблюдается умеренная гипохромная микроцитарная анемия: умеренное снижение гемоглобина при нормальном, а иногда и повышенном количестве эритроцитов, снижение индексов MCV, МСН, МСНС (рис. 89). В мазках крови отмечается анизоци-
тоз, пойкилоцитоз, мишеневидность эритроцитов, может быть базофильная пунктация эритроцитов, выявляется ретикулоцитоз.
В сыворотке крови имеет место умеренная неконъюгированная билиру- бинемия, содержание железа обычно нормальное или повышенное.
Диагноз устанавливается на основании результатов определения малых фракций гемоглобина НЬА2 и HbF. Для больных гетерозиготной формой (3-талассемии характерно повышение содержания фракции НЬА2 до 3,5-8% и примерно у половины больных - HbF до 2,5-7%.
бр-талассемия (HbF-талассемия) представляет собой заболевание, обусловленное нарушением синтеза как Р-, так и 5-цепей глобина. Гетерозиготное состояние характеризуется гипохромной анемией. Содержание НЬА2 нормальное или даже пониженное, содержание HbF значительно повышено и может достигать 5 20%. Клинико-гематологическая картина близка к р-талассемии. Гомозиготная форма 5р-талассемии протекает легче, чем гомозиготная форма р-талассемии. У гомозигот по 5Р-талассемии в крови отсутствуют НЬА и НЬА2, определяется только HbF.
а-талассемия. а-талассемия возникает при делеции генов, кодирующих синтез данной цепи. Продукция a-цепей регулируется двумя парами неодинаковых по значению генов, располагающихся в 11 -й паре хромосом. При дефиците a-цепей в крови новорожденных накапливаются тетрамеры НЬ Bart’s (у4), а в постнатальном периоде (в том числе у взрослых) - Hb Н (Р4). Различают 4 основные формы а-талассемии.
Гомозиготная а-талассемия развивается вследствие полной блокады синтеза a-цепей и характеризуется отсутствием нормальных гемоглобинов (70-100% составляет Hb Bart’s). Hb Bart’s не способен переносить кислород из-за аномально повышенного сродства к нему. Вследствие чего наступает аноксия тканей, приводящая к развитию водянки и внутриутробной гибели плода.
Н-гемоглобинопатия. Обусловлена значительным угнетением продукции a-цепей вследствие отсутствия 3 из 4 генов. Избыточный синтез (3-цепей приводит к их накоплению и образованию тетрамеров |34 (НЬН). У новорожденных 20-40% приходится на долю Hb Bart’s (у4), который позднее меняется на НЬН. Гемоглобин Н в функциональном отношении неполноценен, так как обладает очень высоким сродством к кислороду. НЬН не связывается с гаптоглобином, является нестабильным, нестойким, легко подвергается окислению и осаждается в клетке по мере ее старения. При этом заболевании наблюдается повышенное образование MetHb. Агрегация НЬН вызывает изменение пластичности мембраны эритроцитов, нарушает метаболизм клеток, что сопровождается гемолизом.
Клинически гемоглобинопатия-Н протекает в форме промежуточной талассемии. Заболевание выявляется обычно к концу 1-го года жизни хронической гемолитической анемией умеренной степени тяжести, изредка наблюдается бессимптомное течение. Заболевание характеризуется сравнительно нетяжелым клиническим течением, гепатоспленомегалией, желтушностью, анемией. Изменения скелета незначительные. В костном мозге - умеренная гиперплазия эритроидного ростка, незначительный неэффективный эритропоэз. В крови - выраженная гипохромия и мишене- видность эритроцитов, небольшой ретикулоцитоз. После инкубации крови с крезиловым синим при 55 °С выпадает нестабильный гемоглобин Н в виде множества мелких фиолетово-синих включений в эритроцитах, что отличает ее от других форм а-талассемий. После спленэктомии включения НЬН по внешнему виду начинают напоминать тельца Гейнца. Однако по химической структуре они отличаются от телец Гейнца тем, что состоят из преципитированных p-цепей (Р4), в то время как тельца Гейнца представляют собой осажденные молекулы НЬА (а,Р2) и некоторые другие нестабильные гемоглобины. При электрофорезе сыворотки крови в щелочном буфере наблюдается дополнительная фракция, движущаяся впереди НЬА (быстро- двигающаяся фракция). У взрослых людей значения НЬН составляют 5-30%, до 18% может приходиться на долю Hb Bart’s, НЬА2 снижен (1-2%), HbF в норме или слегка повышен (0,3-3%).
Малая а-талассемия (a-thj). Гетерозиготное состояние по гену a-th,. Синтез a-цепей снижен в умеренной степени. В периферической крови обнаруживаются легкая степень анемии с характерными для талассемии морфологическими изменениями эритроцитов. У новорожденных, носителей этого гена, в пуповинной крови содержание Hb Bait’s не превышает 5-6%. Продолжительность жизни эритроцитов на нижней границе нормы.
Бессимптомная форма а-талассемии (a-th2). Гетерозиготное состояние по гену a-th2 характеризуется незначительным снижением продукции a-цепей, не приводящим к развитию анемии и морфологическим изменениям эритроцитов. У новорожденных, носителей этого гена, Hb Bart’s не превышает 1-2%.
Р-талассемия. Р-талассемия - гетерогенное заболевание. В настоящее время известно более 100 мутаций, вызывающих р-талассемию. Обычно дефект состоит в образовании неполноценной мРНК Р-глобина. Разнообразие молекулярных дефектов приводит к тому, что так называемая гомозиготная Р-талассемия нередко представляет двойное гетерозиготное состояние по разным дефектам синтеза Р-глобина. Различают Р°-талассемию, когда у гомозигот полностью отсутствует синтез p-цепей глобина, и р+-талассемию, при частично сохраненном синтезе p-цепей. Среди р+-талассемий выделяют две основные формы: тяжелую средиземноморскую форму, при которой синтезируется около 10% нормальной цепи (большая талассемия, анемия Кули), и более легкую негритянскую форму, когда сохраняется около 50% синтеза нормальной P-цепи. В г руппу Р-талассемий относят также 5р-талассемию и Hb Lepore. В результате этого имеются существенные расхождения в клинической картине различных форм талассемии, однако для всех р-талассемий общим является внутриклеточный гемолиз эритроцитов, неэффективный эритропоэз в костном мозге и спленомегалия.
Большая талассемия (анемия Кули, thalassemia major). Считается гомозиготной формой талассемии, хотя во многих случаях заболевание является двойным гетерозиготным состоянием по различным формам р-талассемии. Клинически заболевание проявляется к концу 1-2-го года жизни ребенка спленомегалией, желтухой, бледностью кожных покровов, изменением костей - квадратный череп, уплощенная переносица, выступающие скулы, сужение глазных щелей. Дети физически плохо развиты.
В костном мозге наблюдается гиперплазия красного ростка, выявляется значительное количество сидеробластов. В крови - гипохромная микроцитарная анемия (снижены MCV, МСН, МСНС), резкий анизоцитоз, встречаются эритроциты с базофильной пунктацией, эритрокариоциты, пойкилоцитоз, мишеневидные эритроциты, шизоциты (рис. 88). Даже при тяжелой анемии количество ретикулоцитов не бывает высоким, так как в костном мозге выражен неэффективный эритропоэз. Отмечается повышение осмотической резистентности эритроцитов. Характерна лейкопения с относительным лим- фоцитозом, в период гемолитического криза - нейтрофильный лейкоцитоз со сдвигом влево. В период криза может наблюдаться левый сдвиг.
В сыворотке крови имеет место гипербилирубинемия за счет неконъ- югированного билирубина, повышено содержание сывороточного железа. Избыточное отложение железа приводит к сидерозу органов. Характерным
признаком большой талассемии является выраженное увеличение концентрации фетального гемоглобина. Количество НЬА варьирует в зависимости от типа талассемии. У гомозигот с Р°-талассемией НЬА практически отсутствует. При р+-талассемии (средиземноморский тип) НЬА варьирует от 10 до 25%, при Р+-талассемии негритянского типа содержание НЬА значительно выше. Однако тяжесть заболевания не всегда коррелирует с количеством фетального гемоглобина. Содержание НЬА2 может быть различным, чаще повышенным, но отношение НЬА2/ НЬА всегда меньше, чем 1:40. Диагноз подтверждается электрофорезом гемоглобина (уровень HbF - до 70%).
Малая талассемия (thalassemia minor) является гетерозиготной формой Р-талассемии. Клинически малая талассемия характеризуется менее выраженными симптомами, чем большая талассемия, может протекать практически бессимптомно.
В костном мозге - гиперплазия эритроидного ростка, количество си- деробластов повышено или нормальное. В крови наблюдается умеренная гипохромная микроцитарная анемия: умеренное снижение гемоглобина при нормальном, а иногда и повышенном количестве эритроцитов, снижение индексов MCV, МСН, МСНС (рис. 89). В мазках крови отмечается анизоци-
тоз, пойкилоцитоз, мишеневидность эритроцитов, может быть базофильная пунктация эритроцитов, выявляется ретикулоцитоз.
В сыворотке крови имеет место умеренная неконъюгированная билиру- бинемия, содержание железа обычно нормальное или повышенное.
Диагноз устанавливается на основании результатов определения малых фракций гемоглобина НЬА2 и HbF. Для больных гетерозиготной формой (3-талассемии характерно повышение содержания фракции НЬА2 до 3,5-8% и примерно у половины больных - HbF до 2,5-7%.
бр-талассемия (HbF-талассемия) представляет собой заболевание, обусловленное нарушением синтеза как Р-, так и 5-цепей глобина. Гетерозиготное состояние характеризуется гипохромной анемией. Содержание НЬА2 нормальное или даже пониженное, содержание HbF значительно повышено и может достигать 5 20%. Клинико-гематологическая картина близка к р-талассемии. Гомозиготная форма 5р-талассемии протекает легче, чем гомозиготная форма р-талассемии. У гомозигот по 5Р-талассемии в крови отсутствуют НЬА и НЬА2, определяется только HbF.
а-талассемия. а-талассемия возникает при делеции генов, кодирующих синтез данной цепи. Продукция a-цепей регулируется двумя парами неодинаковых по значению генов, располагающихся в 11 -й паре хромосом. При дефиците a-цепей в крови новорожденных накапливаются тетрамеры НЬ Bart’s (у4), а в постнатальном периоде (в том числе у взрослых) - Hb Н (Р4). Различают 4 основные формы а-талассемии.
Гомозиготная а-талассемия развивается вследствие полной блокады синтеза a-цепей и характеризуется отсутствием нормальных гемоглобинов (70-100% составляет Hb Bart’s). Hb Bart’s не способен переносить кислород из-за аномально повышенного сродства к нему. Вследствие чего наступает аноксия тканей, приводящая к развитию водянки и внутриутробной гибели плода.
Н-гемоглобинопатия. Обусловлена значительным угнетением продукции a-цепей вследствие отсутствия 3 из 4 генов. Избыточный синтез (3-цепей приводит к их накоплению и образованию тетрамеров |34 (НЬН). У новорожденных 20-40% приходится на долю Hb Bart’s (у4), который позднее меняется на НЬН. Гемоглобин Н в функциональном отношении неполноценен, так как обладает очень высоким сродством к кислороду. НЬН не связывается с гаптоглобином, является нестабильным, нестойким, легко подвергается окислению и осаждается в клетке по мере ее старения. При этом заболевании наблюдается повышенное образование MetHb. Агрегация НЬН вызывает изменение пластичности мембраны эритроцитов, нарушает метаболизм клеток, что сопровождается гемолизом.
Клинически гемоглобинопатия-Н протекает в форме промежуточной талассемии. Заболевание выявляется обычно к концу 1-го года жизни хронической гемолитической анемией умеренной степени тяжести, изредка наблюдается бессимптомное течение. Заболевание характеризуется сравнительно нетяжелым клиническим течением, гепатоспленомегалией, желтушностью, анемией. Изменения скелета незначительные. В костном мозге - умеренная гиперплазия эритроидного ростка, незначительный неэффективный эритропоэз. В крови - выраженная гипохромия и мишене- видность эритроцитов, небольшой ретикулоцитоз. После инкубации крови с крезиловым синим при 55 °С выпадает нестабильный гемоглобин Н в виде множества мелких фиолетово-синих включений в эритроцитах, что отличает ее от других форм а-талассемий. После спленэктомии включения НЬН по внешнему виду начинают напоминать тельца Гейнца. Однако по химической структуре они отличаются от телец Гейнца тем, что состоят из преципитированных p-цепей (Р4), в то время как тельца Гейнца представляют собой осажденные молекулы НЬА (а,Р2) и некоторые другие нестабильные гемоглобины. При электрофорезе сыворотки крови в щелочном буфере наблюдается дополнительная фракция, движущаяся впереди НЬА (быстро- двигающаяся фракция). У взрослых людей значения НЬН составляют 5-30%, до 18% может приходиться на долю Hb Bart’s, НЬА2 снижен (1-2%), HbF в норме или слегка повышен (0,3-3%).
Малая а-талассемия (a-thj). Гетерозиготное состояние по гену a-th,. Синтез a-цепей снижен в умеренной степени. В периферической крови обнаруживаются легкая степень анемии с характерными для талассемии морфологическими изменениями эритроцитов. У новорожденных, носителей этого гена, в пуповинной крови содержание Hb Bait’s не превышает 5-6%. Продолжительность жизни эритроцитов на нижней границе нормы.
Бессимптомная форма а-талассемии (a-th2). Гетерозиготное состояние по гену a-th2 характеризуется незначительным снижением продукции a-цепей, не приводящим к развитию анемии и морфологическим изменениям эритроцитов. У новорожденных, носителей этого гена, Hb Bart’s не превышает 1-2%.
Источник: Долгов В.В., Луговская С.А., Морозова В.Т., Почтарь М.Е., «Лабораторная диагностика анемий» 2009