АНТИБИОТИКИ, ОБРАЗУЮЩИЕСЯ ПУТЕМ КОНДЕНСАЦИИ НЕСКОЛЬКИХ МЕТАБОЛИТОВ
Синтез многих антибиотиков включает конденсацию двух или более молекул, образующихся в процессе первичного метаболизма, которые могут быть в большей или меньшей степени модифицированы. Из антибиотиков этой группы, используемых в химиотерапии, мы рассмотрим линкомицин и новобиоцин.
- ЛИНКОМИЦИН
Линкомицин образуется путем конденсации циклической аминокислоты с модифицированным углеводом. О биосинтезе компонентов молекулы линкомицина известно сравнительно мало. Показано, что метильные группы при атоме азота, сера и пропильная цепь происходят из метионина. Пирролидиновое кольцо образуется из тирозина в катаболическом пути, который может быть параллелен пути образования меланина. Неполная и во многом гипотетичная схема биосинтеза линкомицина приведена на рис. 6.6.
- НОВОБИОЦИН
Этот антибиотик, по-видимому, синтезируется из трех компонентов: углевода, кумарина и производного бензойной кислоты. Интересно, что как кумарин, так и производное бензойной кислоты происходят от одного первичного метаболита — тирозина. Наиболее вероятная общая схема биосинтеза приведена на рис. 6.7. Порядок, в котором расположены реакции на рисунке, нельзя считать твердо установленным. Для простоты мы показали, что модификации сахара происходят до конденсации с кумарином, но возможно, что метилирование и карбамои- лирование происходят после конденсации. Мы также не знаем точно, на какой стадии включается алифатическая цепь в положение 3 бензольного кольца. Интересно, что эта цепь не образуется из мевалоновой кислоты, что можно предположить, исходя из ее структуры, и, следовательно, ее биосинтез не связан с биосинтезом изопреноидных соединений.
- АНТИБИОТИКИ, ОБРАЗУЮЩИЕСЯ ПУТЕМ
ОЛИГОМЕРИЗАЦИИ ИЛИ ПОЛИМЕРИЗАЦИИ
Процесс биосинтеза антибиотиков, в основе которых лежит структура, образующаяся при конденсации близких мономеров, можно разделить на два этапа: 1) процесс олигомеризации, сходный для всех антибиотиков одной биосинтетической груп-
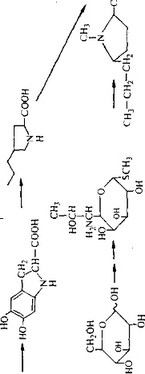
Рис. 6.6. Гипотетический путь биосинтеза линкомицина.
пы, и 2) последующие модификации, характерные для индивидуальных антибиотиков. Эта схема неприменима к аминоглико- зидам, у которых конденсация обычно является последней стадией биосинтеза.
А. ПОЛИПЕПТИДЫ И ДЕПСИПЕПТИДЫ
Имеется около 300 антибиотиков, которые можно считать результатом конденсации аминокислот с образованием пептидных связей. Они отличаются от нормальных белков тем, что
- их мол. масса не превышает 3000; 2) в их состав входят не-
гбычные аминокислоты, например D-аминокислоты, N-метил- ли оксиаминокислоты, р-аминокислоты и т. д.; 3) имеют тен- енцию к циклизации и гиперциклизации.
При конденсации некоторых аминокислот образуются кольца з(например, в случае цистеина — тиазольные кольца), и зачастую молекула представляет собой макроцикл.
I Основной особенностью биогенеза полипептидных антибиотиков является то, что при их синтезе используется специфический ферментный комплекс, хотя долгое время это положение йодвергалось сомнению. Показано, что обычная белоксинтези- ующая система, содержащая комплекс мРНК с рибосомами, е участвует в их синтезе. Первым аргументом в пользу этого ~оложения послужило наличие в пептидных антибиотиках аминокислот, которые отличаются от обычных аминокислот, встречающихся в белках, а также неспособность известных ингибиторов белкового синтеза подавлять биосинтез этих антибиотиков. Позже, когда некоторые из этих антибиотиков были синтезированы в бесклеточных системах, не содержащих ни рибосом, ни РНК, были получены более весомые доказательства. Системы синтеза in vitro позволили идентифицировать основные стадии процесса полимеризации.
- Аминокислоты активируются в реакции с АТР с образованием аминоациладенилатов.
- Высокоэнергетическая связь между аминокислотой и фосфатом адениловой кислоты обеспечивает перенос аминокислоты на тиольные группы в сложной ферментной системе с образованием аминоацилтиоэфиров.
- Ферментная система катализирует затем образование пептидной связи между карбоксильной группой первой аминокислоты и аминогруппой второй аминокислоты, используя энергию расщепления тиоэфирной связи. Цепь, образуемая таким путем, все время остается связанной сложной эфирной связью с тиольной группой, и к ней может присоединяться третья аминокислота, четвертая и т. д. до конца цепи.
Более детально этот процесс описан позже при рассмотрении синтеза грамицидина S.
Важно отметить, что данный механизм синтеза напоминает механизм полимеризации малонатных единиц при синтезе жирных кислот. Принципиальное различие состоит в том, что в системе синтеза полипептидов последовательность аминокислот контролируется, тогда как при синтезе жирных кислот все мономеры идентичны и такой контроль не нужен.
Описанный механизм синтеза объясняет, почему 1) пептидные антибиотики никогда не содержат больше 15 или 20 аминокислотных остатков, поскольку очень маловероятно, что ферментный комплекс может «узнать» большее число аминокислот, и 2) пептидные антибиотики часто образуются «семействами»,
компоненты которых различаются одним или несколькими аминокислотными остатками. Действительно, система узнавания различных аминокислот может ошибаться, а разные аминокислоты могут конкурировать за включение в одно и то же положение.
Очень часто после или во время образования цепи происходят и другие реакции. Это могут быть циклизация молекулы или ее части и модификация отдельных аминокислот. Например, D-аминокислоты, часто встречающиеся в полипептидах, обычно образуются из L-аминокислот, причем показано, что инверсия происходит после полимеризации. Одним из примеров значительной модификации трипептидной цепи служит биосинтез пенициллинов и цефалоспоринов, описанный ниже.
Вероятно, сходные механизмы биосинтеза ответственны за биогенез пептолидов и депсипептидов — антибиотиков, отличающихся от полипептидов наличием вместо некоторых амидных связей сложноэфирных.
- БИОСИНТЕЗ ГРАМИЦИДИНА S
Грамицидин S представляет собой декапептид, состоящий из двух идентичных пентапептидов, циклизованных «голова к хвосту» (рис. 6.8). Биосинтез этого антибиотика катализируется двумя растворимыми ферментами, один из которых имеет
мол. массу 100 000 (легкий фермент, или L), а другой — 280 000 (тяжелый фермент, или Н). Экстракты штамма-проду- цента, Bacillus brevis, содержащие эти ферменты, могут синтезировать антибиотик из аминокислот в присутствии АТР и ионов магния. Легкий фермент катализирует активацию фенилаланина и его переход из левовращающей формы в правовра-
происходит в ходе реакции, представленной на рис. 6.9. После активации пять аминокислот оказываются связанными с ферментами в форме тиоэфиров. Полимеризация осуществляется только в том случае, когда два фермента, несущие активированные аминокислоты, объединены, и протекает так, как показано на рис. 6.10. Как только пентапептиды синтезированы, две • молекулы взаимодействуют с образованием циклического соединения, замыкаемого двумя добавочными пептидными связями лейцин — фенилаланин с расщеплением последних тиоэфирных связей и с освобождением фермента.
- ПЕНИЦИЛЛИНЫ И ЦЕФАЛОСПОРИНЫ
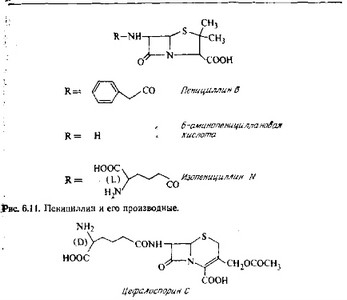
Биосинтез этих антибиотиков исследован главным образом у Penicillium chrysogenum и Cephalosporium acremonium. P. chrysogenum образует различные пенициллины в зависимости от условий ферментации. Некоторые из них, важные с точки зрения биосинтеза, представлены на рис. 6.11. С. acremonium образует пенициллин N и цефалоспорин С, структурные формулы которых приведены на рис. 6.12, а также третий антибиотик, имеющий стероидную структуру.
Образование боковой цепи пенициллина G из фенилуксус- ной кислоты доказывается тем, что 1) при добавлении фенилук- сусной кислоты к культуре P. notatum наблюдается более интенсивное образование пенициллина G и 2) при добавлении радиоактивной фенилуксусной кислоты происходит специфическое включение метки в боковую цепь пенициллина.
Сходные опыты с использованием меченых предшественников показали, что ядро пенициллинов происходит из цистеина и валина. Обе эти аминокислоты включаются в интактной форме. Цистеин остается в левовращающей конфигурации, а валин переходит из лево- в правовращающую форму при асимметрическом центре, но, как показано независимым мечением двух метильных групп, конфигурация при третьем углеродном атоме не изменяется.
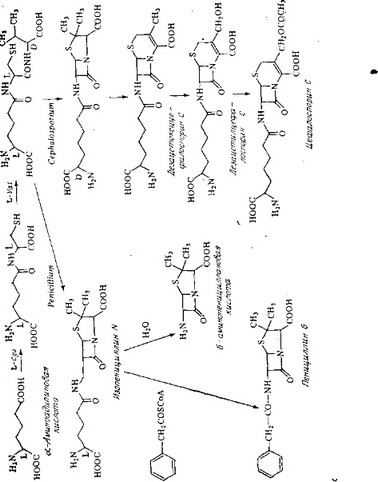
Маловероятно, однако, чтобы в микроорганизме прямо синтезировалось ядро пенициллина (6-аминопенициллановая кислота, 6-АПК) и затем происходила его конденсация с фенилуксусной кислотой. Скорее всего синтезируется промежуточный продукт, трипептид а-аминоадипил-цистеинил-валин, который затем циклизуется с образованием изопенициллина N. Из него в результате обмена боковой цепи может образоваться пенициллин G. Эту гипотезу подтверждают следующие данные.
- Трипептид а-аминоадипил-цистеинил-валин выделен из культуральной жидкости.
H,
 (D) N
(D) N
ноос
- Подавление синтеза а-аминоадипиновой кислоты, например при добавлении лизина, приводит к подавлению синтеза пенициллина G.
- Из Penicillium выделен фермент ацилтрансфераза, катализирующий in vitro следующие реакции:
Изопенициллин N+Фенилацетилкофермент А--gt;-Пенициллин G +
+ lt;х-Аминоадипиновая кислота+Кофермент А,
Изопенициллин Ы+НаО-»-6-аминопенициллановая кислота+ +а-Аминоадипиновая кислота.
Аналогичные опыты с использованием меченых предшественников, проведенные с культурой Cephalosporium, показа
ли, что ядро цефалоспорина и пенициллина N образуется иа левовращающего цистеина и левовращающего валина. У пенициллина N, как и у всех пенициллинов, наблюдается инверсия конфигурации валина при асимметрическом центре. В цефало- споринах одна из двух метильиых групп валина включается в дегидротиазиновое кольцо, а другая вначале гидроксилируется, а затем ацетилируется. Происхождение цефалоспорина С и пенициллина N из трипептида а-аминоадипил-цистеинил-валина с очевидностью следует из их структуры. Более того, этот трипептид был выделен из культуры Cephalosporium. Однако необходимо отметить, что в трипептиде а-аминоадипиновая кислота имеет левую конфигурацию, тогда как в цефалоспорине С и в пенициллине N — правую. Вероятно, в культуре Cephalosporium присутствует фермент, способный катализировать инверсию конфигурации. После такой модификации молекула становится устойчивой к ацилтрансферазам, и, следовательно, боковая цепь в отличие от того, как это происходит в культурах Peniciliium, не может ферментативным путем обмениваться на другие ацильные цепи.
Схема общего пути биосинтеза пенициллинов и цефалоспоринов, приведенная на рис. 6.13, построена с использованием современных данных.
Б. АНТИБИОТИКИ, ОБРАЗУЮЩИЕСЯ ПУТЕМ ПОЛИМЕРИЗАЦИИ
АЦЕТАТНЫХ И ПРОПИОНАТНЫХ ЕДИНИЦ
Из экспериментов с включением меченой уксусной кислоты следует, что многие антибиотики синтезируются путем конденсации ацетатных единиц «голова к хвосту», т. е. с образованием связи между углеродным атомом карбоксильной группы и углеродным атомом метильной группы следующей единицы. По-видимому, иногда структурной единицей вместо ацетата является пропионат, причем в этом случае связь образуется между углеродным атомом карбоксильной группы и метиленовой группой следующей единицы, в результате чего синтезируется метилза- мещенная цепь.
В этой биогенетической группе можно обнаружить совершенно разные структуры: изолированные или конденсированные ароматические кольцевые структуры, хиноны, макролиды, анзамицины. Такое разнообразие является результатом сравнительно небольших вариаций биосинтетического процесса, который в принципе во многом близок биосинтезу жирных кислот.
Чтобы представить себе процесс полимеризации, приводящий к синтезу жирных кислот, рассмотрим в качестве примера биосинтез пальмитиновой кислоты (рис. 6.14). Можно видеть, что уксусная кислота непосредственно участвует только в инициации цепи, а основной мономерной единицей является
малоновая кислота. Однако малоновая кислота образуется из уксусной путем карбоксилирования последней, и поэтому опыты с включением меченых предшественников указывают, что цепь строится исключительно из ацетатных единиц.
Для удлинения цепи как уксусная кислота, так и малоновая должны сначала активироваться. Активация происходит за счет образования тиоэфирной связи с коферментом A (CoASH). Ацетат и малонат благодаря наличию тиоэфирной связи с коферментом переносятся к ферментной системе, осуществляющей полимеризацию и восстановление карбонильных групп с образованием метиленовых групп [путем восстановления в окси- группу, дегидрирования и восстановления двойной связи (не показано на схеме)]. В клетках эукариот эти реакции осуществляются мультиферментным комплексом с очень высокой молекулярной массой (gt;2 000 000), тогда как у бактерий разные реакции катализируются отдельными ферментами.
(1) Лктивация инициатора
amp;J-CO-S — СоА + фермент -SH-^Ri-CO-S
(2} Активация , мономеров
отличие системы синтеза жирных кислот от системы, ответственной за биосинтез вторичных метаболитов, заключается в восстановлении карбонильных групп, которое при синтезе жирных кислот происходит немедленно после конденсации очередной единицы и до начала конденсации следующей. При биосинтезе вторичных метаболитов такого восстановления или не происходит (и тогда образуются, как мы увидим далее, ароматические структуры), или оно осуществляется только частично, до стадии образования гидроксильной
группы или двойных связей, давая начало макролидным структурам.
Другим важным отличием является способность систем, синтезирующих некоторые антибиотики, использовать для элонгации метилмалонат (который может образовываться при кар- боксилировании пропионата) с образованием метилированных цепей. В некоторых случаях используется и этилмалонат, что приводит к образованию этилзамещенных цепей. Аналогично тому, как включение пропионата указывает на то, что цепи синтезируются из метилмалонатных единиц, включение остатков масляной кислоты означает, что промежуточным продуктом является этилмалонат.
Инициатором цепи при синтезе жирных кислот обычно является активированная уксусная кислота, но при синтезе жирных кислот у бактерий это могут быть другие соединения, например пропионат и изовалериановая кислота.
При биосинтезе антибиотиков помимо уксусной и пропионо- вой кислот в качестве инициатора, как мы увидим позже, могут использоваться малонамид и другие более сложные соединения. На рис. 6.15 представлена схема образования цепи при синтезе антибиотиков, происходящих из ацетата и пропионата.
Чтобы связать первичный и вторичный метаболизм, вспомним, что ацетилкофермент А является нормальным продуктом обмена глюкозы; малонилкофермент А также является нормальным продуктом метаболизма, образующимся при карбок- силировании ацетилкофермента А. Аналогичным образом ме- тилмалонилкофермент А может образовываться путем карбок- силирования пропионилкофермента А, но чаще получается с помощью других путей, например при изомеризации сукцинил- кофермента А.
Структура антибиотика определяется главным образом длиной цепи и степенью ее восстановленности. В цепях, образованных исключительно из ацетатных единиц, в том случае, когда карбонильные группы не восстановлены, чередуются метиленовые и карбонильные группы (поликетометиленовые или поли- кетидные цепи). Метиленовые группы в этих структурах высокоактивны и стремятся конденсироваться с карбонильными группами с образованием колец, которые по стерическим причинам содержат преимущественно шесть атомов. Енолизация непрореагировавших карбонильных групп приводит к ароматизации структуры. Некоторые примеры такого рода представлены на рис. 6.16.
Если благодаря частичному восстановлению в процессе полимеризации или в результате наличия метальных заместителей цепь не является истинно поликетометиленовой, циклизация и образование ароматических колец затруднены, и получаются линейные структуры или чаще макроциклы. Классиче-
им примером служат макролиды — как с антибактериальным ействием, имеющие кольца средних размеров, так и противо- рибковые с крупными кольцами (рис. 6.17). Анзамицины ожно рассматривать как промежуточный случай, когда обра- уется и ароматическое кольцо, и макроцикл (рис. 6.17).
Ранее мы подчеркивали, что после образования основы мо- екулярной структуры синтез антибиотика или семейства анти- иотиков обычно завершается в ходе других ферментативных еакций. Мы обсудим примеры таких превращений для некото- ых наиболее важных антибиотиков или в случаях, наиболее нтересных с точки зрения биосинтеза.
- ГРИЗЕОФУЛЬВИН
Биосинтез этого антибиотика в культурах Penicillium griseo- 'fulvum и Penicillium patulum исследовали как с помощью введения меченых предшественников, так и путем выделения предполагаемых промежуточных продуктов. Ранее мы установили, что ;~з одной ацетатной и шести малонатных единиц образуется замещенный бензофенон. Схема вероятной последовательности реакций показана на рис. 6.18. Можно видеть, что синтез идет рт полностью ароматической структуры к частично насыщенной. Именно таким путем, как правило, осуществляется биосинтез вторичных метаболитов из ацетата.
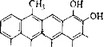
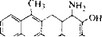
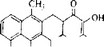
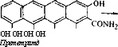
- ТЕТРАЦИКЛИНЫ
Общим предшественником тетрациклина, хлортетрациклина и окситетрациклина является метилпрететрамид (рис. 6.19,Л), получающийся путем конденсации одной малонамидной единицы с восемью малонатными. Метильные группы происходят из
Тон N CONH, 'f' 'Y' 'CONH,
 он он о о OH ОН 0 0
он он о о OH ОН 0 0
г
Рис. 6.19. Биосинтез тетрациклина.
обычного фонда одноуглеродных соединений (фонд доноров метильных групп). Реакции превращения метилпрететрамида в тетрациклин, о существовании которых свидетельствует выделение промежуточных продуктов из блок-мутантов, представлены на рис. 6.19.
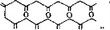
Промежуточные продукты превращения метилпрететрамида в продукт В образуются и при биосинтезе хлортетрациклина. При этом в положение 7 продукта В включается атом хлора, и последующие реакции осуществляются с хлорированным продуктом таким же образом, как и с нехлорированным (рис. 6.20).
Гидроксильная группа в положении 5 окситетрациклина вводится на более поздней стадии, когда ангидротетрациклнн
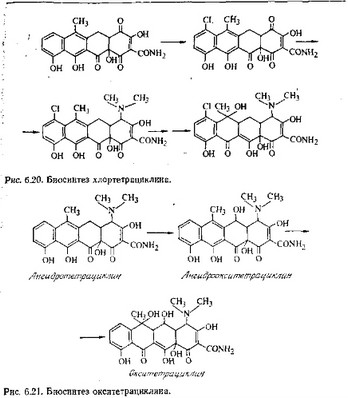
(Г) превращается в ангидроокситетрадиклин (рис. 6.21). Этот продукт претерпевает такие же последующие изменения, как и при биосинтезе тетрациклина.
Если биосинтез происходит в присутствии ингибиторов метилирования, например сульфонамидов, или при использовании мутантного штамма, неспособного к метилированию, вместо ме- тилпрететрамида (Л) образуется прететрамид (рис. 6.22). Последующий биосинтез протекает так же, как описано ранее, и приводит к образованию дезметилтетрациклина (или дезметил- хлортетрациклина). Необходимо отметить, что метилирование азота происходит как обычно, и, следовательно, механизм этого процесса отличается от механизма метилирования углерода.
ОН
conh2
он о он о
Рис. 6.22. Биосинтез 6-дезметилтетрациклина. '
Влияние первичного метаболизма на биосинтез вторичных метаболитов в случае тетрациклинов становится очевидным, когда условия культивирования или введение добавок в среду способствуют превращениям глюкозы по пентозофосфатному пути. Это приводит к усилению биосинтеза антибиотика по сравнению с тем случаем, когда метаболизм глюкозы осуществляется путем гликолиза.
з. ЭРИТРОМИЦИН
У процесса биосинтеза макролидных антибиотиков имеется три аспекта, требующих рассмотрения: 1) происхождение лак- тонного макроцикла; 2) происхождение сахаров; 3) последовательность реакций, приводящих к конденсации этих компонентов, с последующей модификацией.
Мы суммируем здесь все, что известно о биосинтезе эритромицина— наиболее изученного в этом отношении макролидного антибиотика. Как уже отмечалось, основа структуры лактонно- го макроцикла образуется в результате полимеризации одной единицы пропионата и шести метилмалоната.
В разных эритромицинах присутствуют остатки сахаров де- зозамина, микарозы и кладинозы. Установлено, что основа структуры этих сахаров образуется из глюкозы без какого-либо изменения порядка углеродных атомов. Возможная схема их биосинтеза представлена на рис. 6.23.
Дезоксиэритронолид В, единственный выделенный на сегодняшний день промежуточный продукт биосинтеза, путем гид- роксилирования превращается в эритронолид В, который может превращаться либо в эритромицин В в результате конденсации с дезозамином и кладинозой, либо в эритромицин А в результате гидроксилирования по положению 12 и конденсации с дезозамином и кладинозой, либо в эритромицин С путем гидроксилирования по положению 12 и последующей конденса-
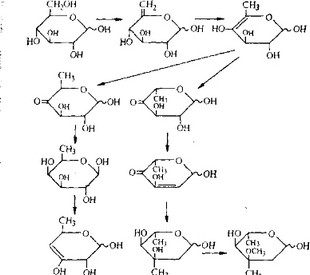
t -микарош L-кладинаэа
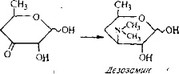
Рис. 6.23. Биосинтез сахаров эритромицина.
ции с дезозамином и микарозой. Вероятно, последовательность этих реакций не является строгой и возможны взаимные превращения некоторых промежуточных метаболитов, так что более сложный продукт, эритромицин А, может образовываться не в одном биосинтетическом пути, а в нескольких, как показано на рис. 6.24.
- РИФАМИЦИНЫ
Основа структуры анзамицинов, содержащих нафталиновое ядро (рифамицин, стрептоварицин, толипомицин, галомицин), берет начало от цепи, образующейся конденсацией двух мало-
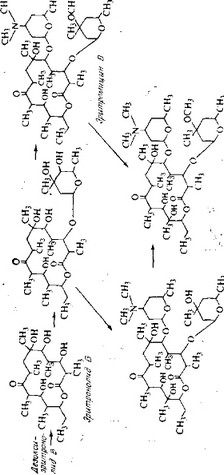
Эритромицин С Эритромицин А Рис. 6.24. Биосинтетические взаимоотношения, характерные для эритромицинов.
натных и восьми метилмалонатных единиц. Характерной особенностью анзамицинов является наличие группировки, ответственной за инициацию синтеза цепи. Недавно было установлено, что это З-амино-5-оксибензойная кислота, которая, как показали опыты по включению 13С-глюкозы, образуется в модифицированном пути биосинтеза ароматических аминокислот. Метилмалонатные единицы могут происходить из пропионовой кислоты, если она добавлена к культуре, но, по-видимому, обычно образуются в результате изомеризации янтарной кислоты.
Характерными особенностями биосинтеза рифамицинов и других анзамицинов является частичная циклизация цепи с образованием второго ароматического кольца, конденсированного с кольцом молекулы-инициатор а, и замыкание макроцикла амидной связью. Структура первых промежуточных продуктов в биосинтезе рифамицинов — проторифамицина и рифамицина W — приведена выше. В последующих реакциях, в частности в результате расщепления алифатической цепи и включения кислорода эфирной группы, рифамицин W превращается в конеч-
ные продукты (рис. 6.25). Главным промежуточным продуктом при этих превращениях является рифамицин S.
Рифамицин В частично превращается в производное с окисленной цепью, рифамицин Y, который не обладает антимикробной активностью. В зависимости от условий культивирования или от используемого штамма рифамицин S может давать начало не только рифамицину В, но и другим продуктам, таким как рифамицин О и рифамицин G, часть структуры которых представлена на рис. 6.26, а также продуктам с неизвестной
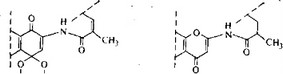
Рис. 6.26. Рифамицин О и рифамицин G (часть структуры; остальная часть молекулы такая же, как у рифамицина В).
CHj—С=0
структурой, в целом называемым рифамициновым комплексом. Другие анзамицины, например толипомицин и галомицин, как считают, должны образовываться из рифамицина W или через рифамицин S, или через сходный промежуточный продукт.
В. ТЕРПЕНОИДНЫЕ АНТИБИОТИКИ
Среди вторичных метаболитов грибов имеется много продуктов с терпеноидной структурой, образующейся конденсацией изопреновых единиц (или, точнее, изопентенилпирофосфатных единиц), за которой часто следует циклизация. Изопентенилпи- рофосфатные единицы синтезируются из ацетата с образованием мевалоновой кислоты в качестве промежуточного продукта (рис. 6.22).
Единственный важный с практической точки зрения антибиотик, образующийся в терпеноидном пути биосинтеза, — фузидиевая кислота, имеющая стероидную структуру. Напомним, что цефалоспорин Р, образуемый Cephalosporium, является стероидом и скорее всего тоже синтезируется по терпе- ноидному пути. По-видимому, биосинтез фузидиевой кислоты осуществляется по стероидному пути, причем в качестве промежуточного продукта образуется сквален. Схема биосинтеза показана на рис. 6.27.
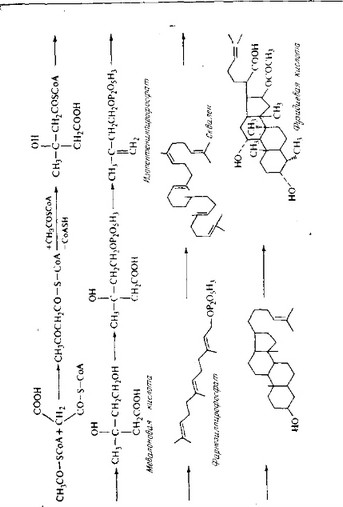
Рис. 6.27. Вероятный путь биосинтеза фузидиевой кислоты.
Г. АМИНОГЛИКОЗИДНЫЕ АНТИБИОТИКИ (АМИНОЦИКЛИТОЛЫ)
Формально аминогликозидные антибиотики следует называть не полимерами, а олигомерами, поскольку они состоят иа немногих мономерных единиц, обычно из трех или четырех. Эти единицы представляют собой циклический многоатомный спирт с шестью углеродными атомами и несколько сахаров, связанных гликозидными связями. Обычно некоторые гидроксильные группы циклитола и сахаров бывают замещены аминогруппой или замещенной аминогруппой, что и дало название этому классу антибиотиков. По некоторым своим особенностям эти структуры напоминают полисахариды бактериальной капсулы и клеточной стенки, например липополисахариды клеточной стенки представителей семейства Enterobacteriaceae с их длинными цепями аминосахаров и аминоспиртов. Можно предположить поэтому, что аминогликозидные антибиотики синтезируются с помощью уже изученных путей биосинтеза бактериальных полисахаридов. Их биосинтез можно разделить на две стадии:
- последовательность реакций, приводящих к превращению глюкозы в индивидуальные звенья, составляющие цепь; 2) полимеризацию этих звеньев путем образования гликозидных связей.
Реакции, в которых один сахар превращается в другой, обычно происходят без перестройки . порядка расположения углеродных атомов. И в циклитолах порядок углеродных атомов такой же, как в молекуле глюкозы. Циклитолы образуются путем замыкания связи между атомами С-1 и С-6. В первичном метаболизме сахара превращаются далее в сахарофосфа- ты или чаще в сахара, этерифицированные в положении 1 ну- клеотиддифоофатом (или дезоксинуклеотиддифосфатом), как в классическом примере превращения UDP-глюкозы в UDP-галактозу. Этерификация в положении 1 нуклеотиддифосфатом является также обычным путем активации сахара, которая необходима для образования гликозидной связи и, следовательно, для олигомеризации или полимеризации.
Мы хотим подчеркнуть, что во всех изученных до сих пор случаях биосинтеза аминогликозидных антибиотиков можно было показать, что индивидуальные единицы образуются из глюкозы без перестройки порядка расположения углеродных атомов. Однако пока не удалось прямо доказать, что гликозид- ная связь образуется после этерификации нуклеотиддифосфа- тами, хотя косвенные данные на этот счет имеются и нет никаких причин предполагать наличие другого механизма.
Главные аминогликозидные антибиотики можно разделить на группы в соответствии со структурой их аминоциклитола (табл. 5.1). Несмотря на структурное сходство, биосинтез разных аминоциклитолов совершенно различен. Например, дезо-
Глюкоэо-б-дюарат v - миоинозит
 ксистрептамин не является предшественником стрептидина, а миоинозит, предшественник стрептидина, не включается в де- зоксистрептамин. Исследован биосинтез лишь немногих амино- гликозидных антибиотиков, а детально прослежен только биосинтез стрептомицина. Мы приведем все известные данные относительно биосинтеза этого антибиотика и отметим несколько моментов, относящихся к биосинтезу неомицина — типичного дезоксистрептаминсодержащего антибиотика.
ксистрептамин не является предшественником стрептидина, а миоинозит, предшественник стрептидина, не включается в де- зоксистрептамин. Исследован биосинтез лишь немногих амино- гликозидных антибиотиков, а детально прослежен только биосинтез стрептомицина. Мы приведем все известные данные относительно биосинтеза этого антибиотика и отметим несколько моментов, относящихся к биосинтезу неомицина — типичного дезоксистрептаминсодержащего антибиотика.
- СТРЕПТОМИЦИН
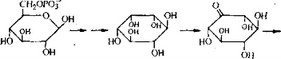
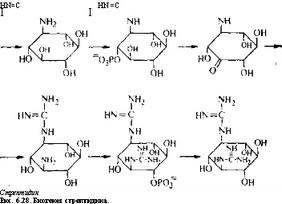
Молекулу стрептомицина можно рассматривать как три- сахарид, образованный из стрептидина (аминоциклитола), L- стрептозы и Ы-метил-Ь-глюкозамина. Лучше всего изучен биосинтез стрептидина. Мы знаем, какие реакции ведут к образованию стрептидина из глюкозы (они показаны в несколько упрощенном виде-на рис. 6.28), а некоторые ферменты, катализирующие эти реакции, очищены и охарактеризованы.
Заметим, что ни стрептамин, ни дезоксистрелтамин не яв- яются промежуточными продуктами этого биосинтетического пути. Гуанидиновые группы образуются в двух разных последовательностях реакций, причем в каждой из этих последовательностей происходит окисление гидроксильной группы в карбонильную, трансаминирование и превращение аминогруппы в гуанидиновую группу в реакции с аргинином. Последней
реакции предшествует фосфорилирование, а сопровождается она гидролизом фосфатной группы.
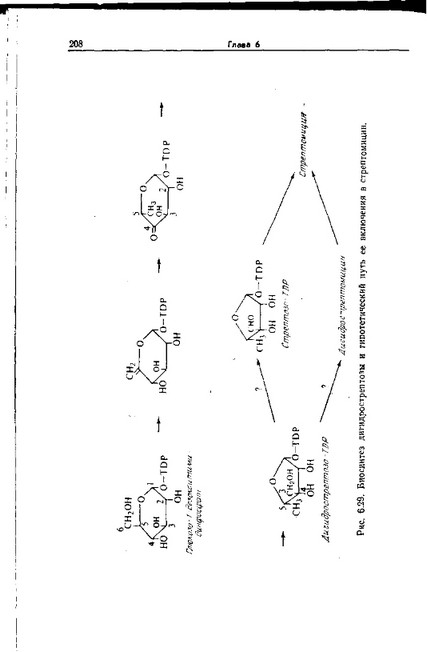
Путь биосинтеза стрептозы окончательно не установлен. Однако нам все-таки известно, что стрептоза образуется из глюкозы путем уменьшения размеров кольца за счет образования связи между атомами С-2 и С-4, тогда как С-3 дает начало альдегидной группе. Недавние исследования показали, что это превращение имеет место после этерификации дезокситими- дин-5'-дифосфатом (dTDP) и что, возможно, не сама стрептоза, а ее предшественник, дигидрострептоза включается в стрептомицин (или, точнее, в дигидрострептомицин) и уже затем последний окисляется (рис. 6.29).
N-метил-Ь-глюкозамин образуется из глюкозы без перестройки расположения углеродных атомов, причем, вероятно, промежуточным продуктом является D-глюкозамин (рис. 6.30). В ходе каких реакций происходит обращение конфигурации — ¦не известно (превращение D-глюкозамина в L-глюкозамин требует эпимеризации четырех асимметрических центров). Мети-
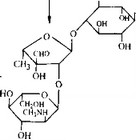
Рис. 6.31. Биосинтез стрептомицина. NuDP — нуклеотиддифосфат, dTDP — дезокситимидиндифосфат.
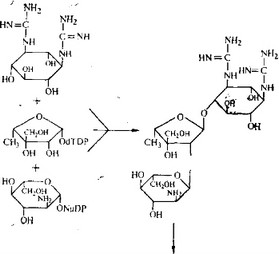 лирование аминогруппы происходит в самом конце, вероятно, после образования гликозидной связи с другими единицами (рис. 6.31). В целом последние стадии биосинтеза стрептомицина, по-видимому, протекают так, как это показано на рис. 6.31.
лирование аминогруппы происходит в самом конце, вероятно, после образования гликозидной связи с другими единицами (рис. 6.31). В целом последние стадии биосинтеза стрептомицина, по-видимому, протекают так, как это показано на рис. 6.31.
- НЕОМИЦИН
Неомицин — это наиболее исследованный из дезоксистреп- таминсодержащих антибиотиков, и тем не менее лишь немногие аспекты его биосинтеза установлены полностью. Как мы уже говорили, дезоксистрептамин отличается от стрептидина по
своему происхождению. Вначале полагали, что он происходит из глюкозамина, но теперь считается, что дезоксистрептамин образуется из глюкозы через производное инозина (рис. 6.32).
Помимо дезоксистрептамина неомицин содержит рибозу и два аминосахара, неозамин В и неозамин С (рис. 6.33). Рибо- за, по-видимому, образуется из глюкозы путем отщепления С-1 в пентозофосфатном пути. Неозамин В и неозамин С образуются из глюкозы через глюкозамин без перестройки расположе-
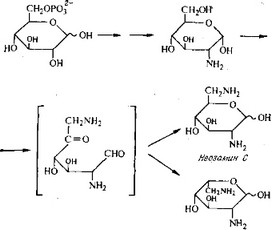
Неоэамин В
Рис. 6.34. Гипотетический путь биосинтеза неозаминов.
ния атомов углерода. Гипотетический путь их биосинтеза представлен на рис. 6.34.
Как происходит сборка компонентов с образованием конечного продукта — неизвестно. Однако выделение ди- и трисаха- ридов из мутантов S. fradiae, микроорганизма, образующего неомицин, и исследование включения предшественников указывают на то, что может иметь место следующая последовательность реакций: 1) неозамин С связывается с дезоксистрептами- ном с образованием неамина; 2) рибоза образует гликозидную связь с неамином, в результате чего получается рибостамицин; 3) последний связывается с неозамином В, и образуется неомицин.
Источник: Ланчини Д., Паренти Ф., «Антибиотики. Пер. с англ. — М.: Мир. — 272 с., ил.» 1985