
Болезнь Фридрейха является наиболее часто встречающейся формой наследственных атаксий. В европейских популяциях распространенность заболевания составляет 2-5 случаев на 100 000 населения, а частота гетерозиготного носительства мутации - около 1 на 100 человек [Harding А., 1984]. Заболевание наследуется по аутосомно-рецессивиому типу и в значительной части семей проявляется в виде единичных случаев. Оба пола заболевают с одинаковой частотой. В типичных случаях болезнь Фридрейха развивается на первом-втором десятилетии жизни и проявляется сочетанием характерных неврологических и экстраневральпых симптомов [Иванова-Смоленская И. А. и др., 1998 (a); Geoffroy G. et al., 1976; Harding А., 1981; 1984]. Неврологическая картина характеризуется смешанной (мозжечково-заднестолбовой) атаксией, дизартрией, сенсорной полиневропатией с выпадением сухожильных рефлексов, парезами и ами- отрофиями, стопными пирамидными знаками и нарушением глубокой чувствительности. К экстраневральным проявлениям болезни Фридрейха, которые в ряде случаев могут предшествовать появлению неврологических расстройств, относятся скелетные деформации (сколиоз, «стопа Фридрейха»), кардиомиопатия, сахарный диабет, гипогонадизм. В последние 20 лет широкое распространение получили «классические» критерии диагноза болезни Фридрейха, предложенные Geoffrey G. с соавторами (1976) и Harding А. (1981). Они включают начало болезни до 20 лет, наличие атаксии, дизартрии, ранней сухожильной арефлексии, симптома Бабинского и одного из указанных выше экстраневральных проявлений.
Патоморфологически болезнь Фридрейха характеризуется гибелью афферентных волокон задних корешков спинного мозга и периферических нервов, а также комбинированной дегенерацией задних и боковых столбов спинного мозга; на поздней стадии процесса дегенеративные изменения обнаруживаются также в ядрах ствола мозга, мозжечке, больших полушариях [Harding А., 1984]. Течение заболевания неуклонно прогрессирующее, больные обычно погибают через 15-20 лет от момента появления первых симптомов.
На рубеже 90-х годов ген болезни Фридрейха был картирован на длинном плече 9-й хромосомы в локусе 9ql3-q21 [Chamberlain S. et al., 1988; Hanauer A. et al., 1990], что дало возможность проведения косвенной ДНК-диагностики заболевания в отягощенных семьях. В нашей стране первый опыт косвенной ДНК-диагнос- тики болезни Фридрейха в ряде информативных семей, основанной на анализе высокополиморфных маркеров из критической хромосомной области, был получен нами совместно с лабораторией ДНК-диагностики Медико
генетического научного центра РАМН [Пугачев В.В. и др., 1996]. В 1996 году большой международной исследовательской группой был идентифицирован новый ген Х25 (другое название данного гена - FRDA), мутации в котором приводят к возникновению болезни Фридрейха [Campuzano V. et al., 1996]. Было показано, что развитие заболевания обусловлено экспансией тринуклеотидных GAA-повторов, локализованных в 1-м нитроне генаХ25. В норме число копий тринуклеотидных GAA-повторов в гене Х25 варьирует от 7 до 22, тогда как у больных болезнью Фридрейха оно составляет от 120 до 1700 [Campuzano V. et al., 1996; Dbrr A. et al., 1996; Filla A. et al., 1996]. У абсолютного большинства больных экспансия GAA-повторов обнаруживается на обеих мутантных хромосомах. В единичных случаях заболевание может вызываться сочетанием экспансии GAA в одном аллеле с наличием точковой мутации во втором аллеле гена Х25 [Campuzano V. et al., 1996; Filla A. et al., 1996; BidichandaniS. et al., 1997], причем хромосомы с точко- выми мутациями составляют не более 2% от общего числа всех мутантных хромосом.
Ген Х25 кодирует белок, состоящий из 210 аминокислот и получивший название «фратаксин». В клетке фратаксин локализуется в митохондриях | Campuzano V. et al., 1997]. В норме наиболее высокая экспрессия фратаксина характерна для мышцы сердца, спинного мозга, скелетной мускулатуры, поджелудочной железы и печени, тогда как у больных болезнью Фридрейха уровень мРНК фратаксина в тканях значительно снижен [Campuzano V. et al., 1996; 1997]. Это свидетельствует о нарушении экспрессии мутантного гена как центральном молекулярном механизме, лежащем в основе развития болезни. Предполагается, что значительное уве- птчение числа копий GAA-повторов в 1-м интроне Х25 ведет к нарушению созревания первичного транскрипта, препятствуя вырезанию интрона из молекулы зрелой мРНК, что приводит к блоку трансляции и отсутствию нормального фратаксина [Rosenberg R., 1996]. Редкие случаи толковых мутаций в кодирующей части гена Х25 ведут к синтезу функционально дефектного фратаксина [Bidichandani S. et al., 1997]. Результаты исследований последних лет свидетельствуют о ключевой роли фратаксина в регуляции митохондриального транспорта железа [Babcock М. et al., 1997; Koutnikova Н. et al., 1997]. Нарушение функции фратаксина в результате мутаций в гене Х25 сопровождается повышением содержания железа в митохондриях, снижением резистентности к ок- сидантному стрессу и окислительным повреждением митохондрий, снижением синтеза АТФ и нарушением энергетического метаболизма клетки [Wilson R., Roof D., 1997; DelatyckiM. etak, 1999; Lodi R.etal., 1999; Wong A. et al., 1999]. Важное значение в патогенезе болезни придается также развивающейся недостаточности Fe-S-за- висимых субъединиц ферментов дыхательной цепи митохондрий [Rotig A. et al., 1997]. Таким образом, с современных позиций болезнь Фридрейха рассматривается как особая разновидность митохондриальной болезни, обусловленная повреждением ядерного гена [Rotig А. et al., 1997]. Не случайно в клинической картине заболевания доминируют симптомы поражения наиболее энергозависимых органов-мишеней (ЦНС, сердце, скелетные мышцы, поджелудочная железа).
Прямая ДНК-диагностика болезни Фридрейха основана на ПЦР-амплификации тринуклеотидного участка гена Х25. При электрофорезе продуктов амплификации мутантные ал лежи определяются как фрагменты ДНК увеличенной длины, нередко в виде смазанного пятна (последнее обусловлено соматическим мозаициз- мом мутантного GAA-повтора и особенностями амплификации сверхдлинных тринуклеотид-содержащих аллелей) (рис. 64). В типичных случаях у больных отсутствуют аллели нормальной длины. Эффективность амплификации значительно снижается по мере нарастания числа триплетов, поэтому аллели с наибольшей степенью экспансии повторов (свыше 1000 копий) на электрофоре! рамме могут отчетливо не визуализироваться. У 1 аких больных для прямой ДНК-диагностики применяется более грудоемкий метод блог -гибридизации [Durr А. fetal., 1996].
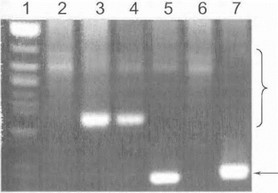
1*ис. 64.11рям?1я ДНК-диагностика болезни Фридрейха Дорожка 1 маркер. Дорожки 2 4 и 6 - больные с болечпыо Фридрейха, дорожка 5 - гетерозиготный носитель мутации, дорожка 7 - контроль. Область мутантных аллелей о различной степенью экспансии GAA-повтОров обозначена фигурной скобкой, нормальный аллель указан стрелкой.
Некоторые лаборатории предпочитают использовать блот-i ибридизацию дня рутинной ДНК-диагности- 1си болезни Фридрейха. В редких случаях, когда больной является компаунд-гетерозиготой но экспансии GAA- повторов в одном аллеле и точковой мутации в другом аллеле гена, на электрофореграмме будут видны 2 продукта амплификации - увеличенной (экспансия повторов) и нормальной длины. В такой ситуации сам по себе факт обнаружения одного патологически удлиненного аллеля при наличии соответствующей клинической картины позволяет с высокой вероятностью склониться в пользу диагноза болезни Фридрейха, однако окончательное подтверждение возможно только после выявления точковой мутации на второй хромосоме, которое проводится с помощью стандартных подходов (чаще всего используется SSCP-анализ и секвенирование кодипую- щей области гена).
В 1997-2000 гг. нами были обследованы 28 больных из 20 семей, поступивших в нейрогенетическое отделение НИИ неврологии РАМН с подозрением на болезнь Фридрейха. При проведении прямой ДНК-диаг- ностики экспансия тринуклеотидных GAA-повторов в 1-м интроне гена Х25 была выявлена у больных из 15 семей, что явилось молекулярным подтверждением диагноза болезни Фридрейха (см. рис. 64). У 5 больных (все - изолированные случаи) результат ДНК-диагностики оказался отрицательным, и таким образом данный диагноз был исключен. Число GAA-повторов у больных варьировало в достаточно широких пределах - от 100 до 1200 (680 ± 350), тогда как в норме во всех случаях оно было менее 50 повторов, что вполне соответствует данным других авторов. Нами не было выявлено каких- либо различий в распределении повторов среди больных различных этнических групп. Распределение мутантных аллелей генаХ25 по числу тринуклеотидных GAA- повторов, выявленное в нашем исследовании, представлено на рис. 65. У 19 больных из 14 семей выявлена эк
спансия GAA-повторов в гомозиготном состоянии. В одной семье у 4 больных сибсов, наряду с экспансией GAA-повторов на одной из хромосом, был выявлен также аллель с нормальной длиной повтора, что позволяет предполагать у них наличие точковой мутации гена на второй мутантной хромосоме. Таким образом, в нашей выборке больных свыше 96% не связанных общим происхождением мутантных хромосом характеризовались экспансией GAA-повторов в соответствующем участке гена Х25.
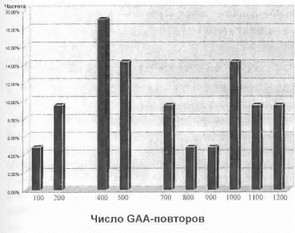
Рис. 65. Распределение мутантных аллелей гена Х25 по числу GAA-повторов в российской популяции
Все больные с «классическим» фенотипом болезни Фридрейха, характеризовавшимся началом болезни Ю 20 -цех, быстрым прогрессированием и сочетанием типичных неврологических и экстраневральных проявлений, имели гомозиготпую экспансию GAA-повторов. При этом число повторов у них в одном или (чаще) обоих аллелях гена было значительным и всегда превышало 400 копий. У больных из 2 семей, имевших небольшую степень экспансии GAA-повторов в обоих аллелях гена (от 200 до 500 копий), клиническая картина была весьма атипичной: она характеризовалась необычно поздним началом (22-48 лет]| относительно медленным течением (на протяжении многих десятилетий), а также отсутствием в клинической картине таких «облигатных» для болезни Фридрейха проявлений как симптом Бабин- ского, кардиомиопатия, скелетные деформации [Илла- риошкин С.Н. и др., 1999 (а, б)]. Аналогичный «мягкий» вариант болезни с необычно медленным прогрессированием и сохранностью мышцы сердца наблюдался нами у больных, являвшихся компаунд-гетерозиготами по экспансии GAA-повторов и точковой мутации в гене Х25. Еще у одного больного, имевшего экспансию GAA-повторов на обеих мутантных хромосомах, клиническая картина характеризовалась крайне необычным для болезни Фридрейха синдромом - спастической атаксией [Иллариошкин С.Н. и др., 2000]. Приводим краткую выпирку из истории болезни этого пациента, демонстрирующую решающую роль прямого ДНК-анализа в постановке диагноза атипичных случаев болезни Фридрейха.
Больной Ф.А., 30 лет, в возрасте 14 лет впервые стал отмечать ограничение движений в стопах, утомляемость в ногах при ходьбе. С 16 лет изменился почерк, появились жалобы на «перебои» в работе сердца. До 18 лет заметного прогрессирования болезни не отмечалось. В 1986 году во время службы в армии наросла слабость в ногах, изменилась походка, в это же время больной стал ощугцатъ скованность в носах, усиливающуюся при длительном беле и ходьбе. В 1988 году обследован в военном госпитале, где впервые отмечена Реформация стоп, повышение тонуса в ногах, атаксия, изменения на ЭКГ. В дальнейшем постепенно нарастала вырйamp;Ш*тость атаксии, развился нижний спастический парапарез, с 23 лет стала невозможной самостоятельная ходьба, ухудшилась речь. Неоднократно находился на обследовании в неврологических стационарах с диагнозами «атипичная форма болезни Фридрейха со спастическим парапарезом.», «наследственная спастическая атаксия», «осложненная форма наследственной спастической параплегии». В 1998 году поступил в нейрогенетическое отделение НИИ неврологии РАМН для уточнения диагноза.
Общий и семейный анамнез не отягощен. При поступлении левосторонний сколиоз грудного отдела позвоночника, деформаъщя стоп по типу «фридрейхов- ских». В неврологическом статусе отмечаются слабость конвергенции и горизонтальный нистагм; диафония; речь растянутая, дизартрия!шя. Снижен темп движений в дистальных отделах рук; легкая, гипотрофия и снижение силы межкостных мышц кистей и .мышц области hypothenar Выраженный нижний парапарез с резким повышением мышечного тонуса по •спастичесому типу в четырехглавои мышце бедра с двух сторон и приводящих группах мышц. В голеностопных суставах движения практически отсутствуют, контрактуры; в положении лежа возможно ограниченное сгибание ног в коленных суставах и кратковременное отрывание пятки от постели. Сухоэюилъные и периостальные рефлексы с рук оживлены, выявляются симптомы Якобсона—Ласка Тремнера, Гофмана С обеих сторон. Коленный и ахиллов рефлексы высокие, поликинетичные, с расширением рефлексогенных зон, выявляются 2-сторонние симптомы Бабанского, Рос- солимо, Оппензейма, отмечается защитная реакция в виде тройного сгибания нижних конечностей в ответ на укол. 1 иперметрия и адиадохокинез в руках, мимопо- падание при палъцеиосовой пробе с двух сторон; почерк неровный, изменен по типу макрографии. Координатор- ные пробы в ногах проверить не удается из-за спастического парапареза. Отчетливая атаксия туловища в положении сидя; стоять не может далее с поддержкой. Болевая и тактильная чувствительность в конем ностях снижена по дистальному типу (в ногах до уровня коленного сустава и в руках до уровня верхней трети предплечий); суставно-мышечное чувство нарушено в пальцах стоп, вибрационная чувствительность снижена в дистальных отделах конечностей.
Клинические анализы крови и мочи, а также рутинные биохимические показатели (включая уровень глюкозы крови) в пределах нормы. На ЭКГ выявлены диффузные изменения миокарда боковой стенки левого желудочка (миокардиодистрофия). Анализ данных, полученных при электронейромиографии, свидетельствует о поралсетш сенсорных волокон периферических нервов (преимущественно на ногах), при сохранном проведении возбуждения по двигательным волокнам. При исследовании соматосенсорных вызванных потенциалов определяется гмубое нарушение функции соматосенсорных периферических путей на участке «срединный нерв—плечевое сплетение» с обеих сторон. .
ДНК-диагностика: при исследовании области тринуклеотидных GAA-повторов в гене Х25 выявлено увеличение длины амплифшщрованного фрагмента в обоих аллелях (число GAA-повторов составило 600 и 700 копий). Клинический диагноз: болезнь Фридрейха.
Взаимоотношению между характером генетического дефекта и особенностями клинической картины
при болезни Фридрейха посвящено большое число исследований. Наши собственные данные и аналогичные данные других авторов показывают, что с нарастанием числа GAA-повторов мутантного гена имеет место бо- Лее ранняя манифеетация болезни [Иллариошкин С.Н.,
1997; И'ллариошкин С.Н. и др., 1999 (а, б); Durr A. et al., 1996; Filla A. et al.. 1996]. Более того, такие тяжелые проявления болезни Фридрейха, как кардиомиопатия и сахарный диабет, развиваются только у тех больных, число GAA-повторов у которых превышает 800 копий | Filla A. et al., 1996]. С другой стороны, при небольшой степени экспансии GAA-повторов могут иметь место разнообразные атипичные, «доброкачественные» варианты болезйи - такие как болезнь Фридрейха с поздним началом (на 5-м и даже 6-м десятилетии жизни!), болезнь Фридрейха с сохранными сухожильными рефлексами или со спастичностыо (что обусловлено меньшей выраженностю сенсорной полиневропатии) и др. [Durr A. et al., 1996; Ragno М. et al., 1997; Coppola G. et al., Г999]. Взаимосвязь между числом триплетов и тяжес- ью заболевания наиболее четко выражена меньшего из двух мутантных аллелей, поскольку он является более «сохранным» с функциональной точки зрения и непосредственно определяет количество активного фра- таксина в клетке [Filla A. et al.. 1996]. Характер проявления толковых мутаций в гене Х25 зависит от влияния мутации па жспрессию гена; миссенс-мутации, при которых фратаксии сохраняет определенный уровень остаточной активности, проявляются «мягким» фенотипом болезни; в то же время повреждения, ведущие к угнетению синтеза белка (нонсенс-мутании, мутации в сайтах сплайсинга), могут характеризоваться «классическим» тяжелым вариантом заболевания с ранним началом симптомов [Filla A. et al., 1996; Bidichandani S. et El., 1997; Forrest S. et al., 1998; Cossee M. et al., 1999].
Таким образом, в настоящее время стало очевидным, что истинный спектр клинических проявлений болезни Фридрейха гораздо шире, чем это предполагалось до введения в практику методов прямого ДНК-тестиро- вания. В целом, по данным ряда авторов, около 30-40% больных с экспансией GAA-повторов в гене Х25 характеризуются теми или иными отклонениями от «классических» критериев болезни Фридрейха [Campuzano V. et al., 1996; Durr A. et al., 1996; Filla A. et al., 1996], в связи с чем многие случаи заболевания оставались до последнего времени не диагностированными. Это позволяет пересмотреть в сторону увеличения данные об истинной распространенности болезни Фридрейха. Можно заключить, что исследование гена Х25 показано во всех аутосомно-рецессивных и спорадических случаях атаксий дегенеративной природы (независимо от возраста дебюта заболевания), особенно при наличии в клинической картине хотя бы отдельных симптомов, свойственных типичному фенотипу болезни Фридрейха (например, кардиомиопатии, нарушений глубокой чувствительности и т.д.).
В настоящее время выявление с помощью ДНК- диагностики случаев болезни Фридрейха на ранней и пресимптоматической стадии приобретает особое значение в связи с потенциальными возможностями превентивной терапии, перспективы которой становятся все более реальными в свете новых знаний о патогенезе болезни. Первые подходы к патогенетической терапии болезни Фридрейха уже сейчас оживленно обсуждаются в литературе (применение десферала и других хелатных соединений с целью коррекции нарушенного митохондриального транспорта железа, назначение коэнзима Q и всего комплекса препаратов, используемых при лечении митохондриальных болезней, и др.) [Cossee М. et al., 1997]. По-видимому, в ближайшем будущем можно ожидать появления этих и целого ряда других принципиально новых методов лечения болезни Фридрейха в арсенале практических врачей-неврологов.