
При всех формах аутосомно-доминантных атаксий с идентифицированными генами (СЦА типа 1, 2, 3, 6, 7, 8, 10, 12, 17 и ДРПЛА) возможно проведение прямой ДНК-диагностики, основанной на амплификации тринуклеотидного (при СЦА10 пентануклеотидного) участка гена. При электрофорезе продуктов амплификации мутантный аллель четко определяется как патоло
гически удлиненный фрагмент ДНК, содержащий увеличенное (надпороговое) число тринуклеотидных повторов. Для: большинства форм атаксий различия между верхней границей нормальных аллелей и нижней границей мутантных аллелей составляют как минимум несколько триплетов (9-12 нуклеотидов и более), в связи с чем качественное электрофоретическое разделение нормальных и мутантных аллелей может проводиться по упрощенному эка ipecc-лротоколу с использованием агарозного геля (рис. 61).
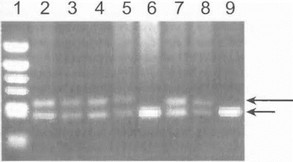
Рис. 61. Прямая ДНК-Д1 [агностика (’ЦА1 (электрофорез в агарозном геле)
Дорожка 1 маркер, дорожки S, 3,5.7— больные ГЦАI, дорожки 4 и 8-носители мутантного гена на прсеьмп гома. лческоп стадии, дорожка 6 родственник из группы риска, у которого носа, гелъamp;тво мутация отвергнуто, дорсискч-i 9-здоровый контроль. Длин «nil стрелкой указан мутан i пый аллель (экспансия CAG-пов i оров гена С ЦА1). короткой стрелкой - нормальный! аллель.
При необходимости точного количественного определения числа тринуклеотидных повторов разделение продуктов амплификации проводится в полиакриламид- | юм геле, обладающем более высокой разрешающей способностью (альтернативой является использование осо-
бой высокоразрешающей агарозы). Это требуется, в частности, при проведении ДНК-диагностики СЦА6, поскольку при данной форме аутосомно-доминантной атаксии разница между нормальными (lt;18 С AG-повторов) и мутантными аллелями (gt;21 CAG-повторов) является весьма небольшой и требует точной количественной оценки длины амплифицированных фрагментов гена. В редких случаях при выраженной экспансии тринуклео- тидных повторов (более 100 копий) ПЦР-диагностика болезни не представляется возможной; это обусловлено весьма низкой эффективностью амплификации аллелей, содержащих сверхдлинные тринуклеотидные тракты. Такая проблема обычно возникает при анализе гена в ювенильных и ранне-детских случаях доминантных атаксий (особенно СЦА7), обусловленных крайней степенью тяжести мутации. Выявление указанных сверхдлинных аллелей проводится путем блот-гибридизации геномной ДНК с (САйД-зондами.
Серьезной проблемой при прямой ДНК-диагно- стике аутосомно-доминантных атаксий является генетическая гетерогенность данных заболеваний и необходимость «перебора» большого числа генов для выявления мутации в одном из них у конкретного больного. Следовательно, разработка рационального алгоритма генетического скрининга представляет собой важнейшую задачу при обследовании больных и семей с аутосомно- доминантными атаксиями. Такой алгоритм может базироваться на двух основных принципах - анализе клинической картины и сравнительной частоте различных форм атаксий в изучаемой популяции. Те или иные особенности клинического синдрома могут позволить заподозрить конкретную генетическую форму атаксии и начать поиск мутации с анализа соответствующего гена. Так, наличие у больного с доминантной атаксией экст- рапирамидной симптоматики позволяет с высокой вероятностью предположить экспансию CAG-повторов в гене СЦАЗ (болезнь Мачадо-Джозеф), сочетание атаксии с дегенерацией сетчатки требует в первую очередь исследования гена СЦА7 и т.д. Как указывалось выше, однако, большинство случаев аутосомно-доминантных атаксий характеризуются отсутствием каких-либо пато- гномоничных симптомокомплексов, которые могли бы служить надежными опорными пунктами для выбора «пужного» гена. В такой ситуации последовательность анализа генов определяется частотой отдельных генетических форм атаксий. Распределение аутосомно-доми- иантных атаксий на генетические формы чрезвычайно варьирует в различных странах и географических регионах, однако в целом свыше 70% данных заболеваний в изученных популяциях мира приходятся на СЦА1, СЦА2, СЦАЗ и СЦА6. В Северной Америке, Японии, Китае и ряде европейских стран (Германия, Португалия, Франция) преобладающей формой является СЦАЗ/болезнь Мачадо-Джозеф - от 21% до 50% случаев атаксий | Maruyama Н. et al., 1995; Ran am L. et al., 1995; Schuls L. et al., 1995; Silveira I. et al., 1996; Tang B. et al., 2000], на Кубе и в Южной Корее ведущей формой является СЦА2 [Gispert S. et al., 1993; Kyu Jin D. et al., 1999], тогда как в Италии и Великобритании превалирует СЦА1 - от 26% до 51% всех семей с аутоеомио-доминантными атаксиями [Giunti R et al., .1994; Pareyson D. et al., 1999]. Таким образом, в каждой конкретной популяции у обследуемых больных с аутосомно-доминантными атаксиями ДНК-диагностику следует начинать с анализа тех генов, мутации в которых являются наиболее частыми в данной популяции.
Предполагается, что различия в частоте отдельных форм атаксий в разных популяциях могут быть связаны с некоторыми молекулярными факторами - например, определенными межпопуляционными различиями
в распределении числа повторов нормальных аллелей, являющихся источником мутаций de novo; большое значение придается также историческим и географическим особенностям возникновения и распространения отдельных мутаций. Так например предполагается, что СЦАЗ/ болезнь Мачадо-Джозеф в большинстве семей Северной Америки, Японии и Австралии обязана своим происхождением большим оригинальным семьям португальских островов Флорес и Сан-Мигель, откуда заболевание было распространено в эти страны португальскими мореплавателями в XVII веке [Rosenberg R., 1992; Burt Т. et al., 1996].
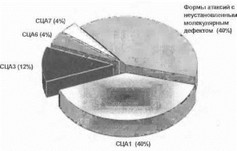
Рис. 62. Частота основных форм аутосомно-доминантных атаксий в российской популяции
Нами в 1994-1999 гг. проведен молекулярно-генетический скрининг больных с аутосомно-домииант- ными атаксиями в российской популяции, позволивший установить частоту основных молекулярных форм этих заболеваний в нашей стране (рис. 62). В нашей выборке в 10 из 25 обследованных неродственных семей (40% семей) заболевание было обусловлено экспансией CAG- повторов в гене СЦА1. В небольшом числе семей с ауто-
сомно-доминантными атаксиями нами были выявлены мутации в генах СЦА2, СЦАЗ, СЦА6 и СЦА7 (рис. 63). В 44% семей у больных были выявлены нормальные значения числа тричуклеотидныл повторов в исследуемых генах. Это позволяет заключить, что в данных случаях заболевание обусловлено повреждением других генов аутосомно-доминантных атаксий (например - СЦА4, СЦА5, СЦА16 и т.д.), непосредственный мутационный анализ которых в настоящее время невозможен. Результаты наших исследований показывают, что в российской популяции прямая ДНК-диагностика аутосомно-доминантных атаксий должна начинаться с анализа (CAG)n- повтора в гене СЦА1 на хромосоме 6р22-23 [Иллари- ошкин С.Н. и др., 1996 (б); 1999 (б)].
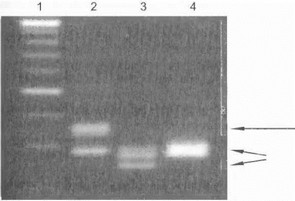
Рис. 63. Прямая ДНК-диагностика СЦА6 Дорожка 1 - ма pfcep, дорожка 2 - болы юй СЦА6, дорожка 3 - бон ьвой с другой‘генетической формой а^тосомно-ДОминантной атаксии, дорожка 4 здоровый контроль. Мутантны|К|Илель сажспансией CAG-повторов указан длинной стрелкой, норМЙЛьные аллели - короткими стрелками. Цректрофорек продуктов амплификации I |ровэден с использованием выс-окоразрешающеййгарозы MS (2Дgt;%).
В обследованных нами семьях с СЦА1 в б случа ях оказалось возможным проведение прямой ДНК-ди- агностики носительства мутантного гена у клинически здоровых родственников из группы риска - детей больных СЦА1. В 4 случаях было диагностировано пресим- нтоматическое носительство мутации; у 2 тестируемых лиц результат ДНК-диагностики был отрицательным, и носительство мутантного гена отвергнуто (см. рис. 61) [Иллариошкин С.Н. и др., 1997]. Лица, являющиеся носителями мутантного гена, были взяты под динамическое наблюдение с целью оказания им помощи в решении ряда психологических, медицинских и социальных проблем.
Нами были обследованы свыше 40 больных с различными спорадическими формами атрофий мозжечка, и ни в одном случае у них не было выявлено мутаций в изучаемых генах. Это совпадает с результатами других авторов [Куй Ли D. et al., 1999; Pujana М. et al., 1999] и свидетельствует о том, что новые мутации в рассматриваемых генах представляют собой исключительную редкость. Можно заключить, что при отсутствии у больного с мозжечковой атаксией семейного анамнеза вероятность обнаружения мутации в одном из генов аутосом- но-доминантных атаксий крайне мала, и проведение ДНК-диагностики у такого больного с практической точки зрения нецелесообразно.
Как указывалось выше, для 6 форм аутосомно- доминантных атаксий известна хромосомная локализа ция мутантного гена, однако сам ген и мутации в нем пока не установлены (см. таблицу 6). В настоящее время оценить частоту данных форм атаксий не представляется возможным, поскольку их диагностика требуй! подтверждения генетического сцепления с соответству ющими хромосомными локусами, что выполнимо ЛИП 111 в редко встречающихся больших информативных родословных. Нри обследовании таких больших семей ДНК- анализ проводится в 2 этапа: 1) скриншг известных мутаций (экспансия тандемных повторов) в генах СЦА-1, 2, 3, 6, 7, 8, 10, 12, 17 и ДРПЛА; 2) при исключении данных мутаций в идентифицированных генах проводится анализ сцепления с маркерами остальных локусов аутосомно-доминантных атаксий - на хромосомах 16q22.1 (СЦА4), llql3 (СЦА5), 15ql4-21.3 (СЦА11), 19ql3.3—13.4 (СЦА13), 19ql3.4-qter (СЦА14) и 8q22.1- 24.1 (СЦА16). При подтверждении сцепления с одним из локусов в обследуемой семье может проводиться косвенная ДНК-диагностика болезни у лиц из группы риска.