Глава 34 ЭПИДЕМИОЛОГИЧЕСКАЯ ХАРАКТЕРИСТИКА ГЕНЕТИЧЕСКИ ОБУСЛОВЛЕННЫХ ЗАБОЛЕВАНИЙ
Генетически обусловленные заболевания представляют собой классический пример вертикальной передачи наследственного материала, который обеспечивает здоровье или развитие различных патологических состояний. Именно к генетически детерминированным заболеваниям наиболее применимо понятие вертикального механизма передачи как эволюционно-обусловленного, обязательного, а не случайного. Все качества (свойства), определяющие индивидуальные колебания в пределах «нормы», генетически детерминированы. Например, устойчивость к инфекции за счет неиссякаемых факторов защиты, степени полноценности иммунного ответа.
В данном разделе представлен эпидемиологический подход изучения генетически обусловленных заболеваний и наследственной предрасположенности к той или иной патологии.
Эпидемиология генетически обусловленных заболеваний не только требует изучения их на молекулярном уровне с использованием специфических генетических приемов исследования, но и популяционных исследований на базе принципов эпидемиологической диагностики.
 |
Эпидемиологическая диагностика лежит в основе изучения причин и закономерностей распространения генетических факторов и наследственной предрасположенности возникновения патологии. Предметом этих исследований является изучение причин возникновения и закономерностей развития наследственных болезней и факторов наследственной предрасположенности, зависящих как от определенных биологических (генетических), так и от природных, и социальных условий. Эпидемиологические методы представляют собой совокупность приемов, которые позволяют выявить и изучить конкретные факторы риска развития и распространения наследственно передающихся форм патологии, установить механизм их действия, оценить и выявить причины того или иного заболевания и разработать систему профилактики.
Определений
Наследственными заболеваниями являются те патологические состояния, в этиологии которых ведущую роль играет генетический компонент. Все патологические состояния имеют тот или иной наследственный вклад, однако в зависимости от степени этого вклада происходит разделение на моногенные (монофакторные) наследственные заболевания (менде- лирующие), для которых определяющим фактором являются генетические нарушения, и полигенные (или мультифакториальные) заболевания, в этиологии которых основное значение принадлежит различным факторам экзогенной природы. Генетические факторы играют одну из ключевых ролей в возникновении и распространении в популяции патологических процессов и состояний, однако конкретный вклад наследственных факторов в возникновение той или иной нозологической формы различен.
Множество заболеваний развиваются в результате влияния внешних повреждающих факторов (в том числе экологических) на фоне наследственной предрасположенности. Существует также и третья группа заболеваний, в этиологии которых решающая роль принадлежит различным экзогенным факторам (например, инфекции или травмы). При этих заболеваниях роль генотипа ограничена регулированием степени восприимчивости организма, эффективностью иммунного ответа и возможностями адаптационно-компенсаторных реакций в ответ на внешнее воздействие.
Врожденные патологические процессы могут быть как генетически детерминированными, так и внутриутробно приобретенными. Генетически детерминированные врожденные заболевания развиваются в результате повреждения генетического аппарата родителей, имеют выраженный наследственный характер и наследуются по доминатному, рецессивному или смешанному типу. Генетически детерминированные болезни могут также быть результатом мутации «de novo» — вновь возникающих мутаций в гаметах родителей. Внутриутробно приобретенные болезни являются врожденными состояниями. Они возникают в результате действия мутагенов в период беременности или связаны с наличием патологии у матери и особенностями внутриутробного развития плода. Помимо сре- довых факторов риска, генетические, наследственные факторы определяют степень возможности возникновения любых форм заболеваний в популяции человека. Классификация болезней человека, выработанная на основе изучения степени влияния наследственных и средовых факторов, дает представление о размере вклада наследственных и средовых факторов риска в развитие различных форм патологических состояний. Ниже представлена структура патологии человека (табл. 34.1).
| Часть IV. ЭПИДЕМИОЛОГИЯ ГЕНЕТИЧЕСКИ ОБУСЛОВЛЕННЫХ ЗАБОЛЕВАНИЙ
Таблица 34.1 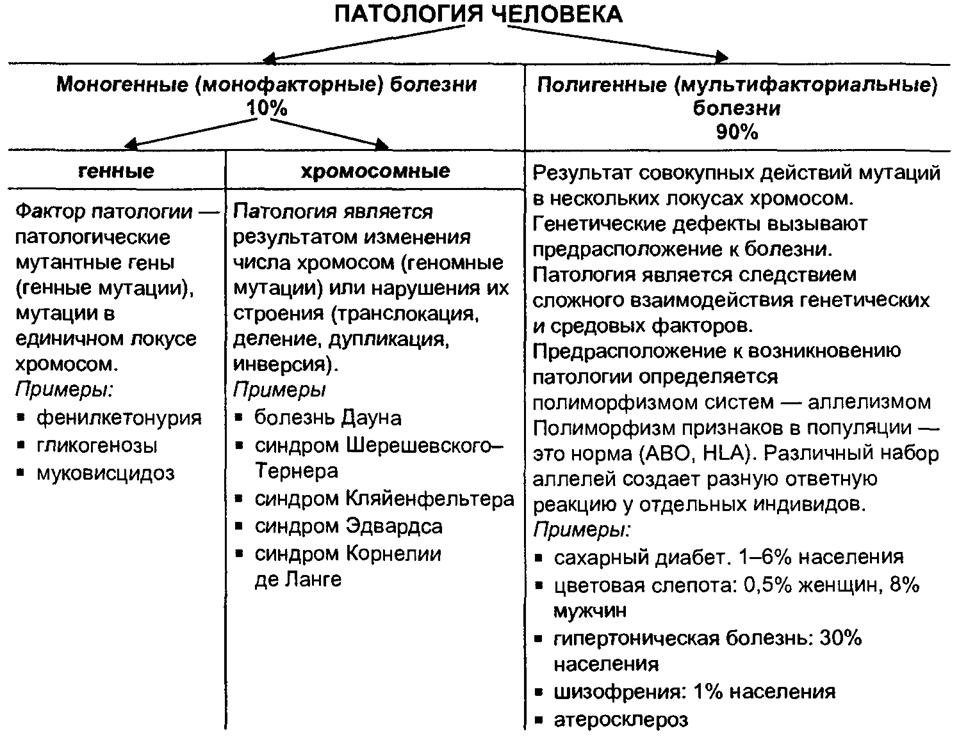 |
Из приведенной классификации видно, что около 10% всех форм патологических состояний являются монофакторными болезнями, причина которых исключительно генетическая — поломки на генном или хромо- сомальном уровне. Функциональная единица наследственности — ген. Через половые клетки родителей передаются не признаки, а информация о них. Первичное действие генов состоит в том, что они программируют биосинтез ферментов по принципу «один ген — один фермент». Ферментные системы контролируются соответствующими комплексами генов и изменения (мутации) в них гена влекут за собой цепи процессов — изменяется или выпадает фермент, что приводит к выпадению соответствующей ступени метаболической реакции и, как следствие, к изменению или нарушению развития отдельных признаков организма, т. е. развитие наследственных признаков идет по схеме «ген — фермент — биохимическая реакция — признак». Для моногенных заболеваний характерно проявление признака в альтернативной форме: есть генетическая поломка — есть болезнь, например фенилкетонурия (ФКУ), нет поломки (дефекта генома) — нет болезни. Тогда как у полигенных заболеваний признак варьируется количественно (например, такой признак, как артериальное давление, есть у всех особей, но уровень проявления этого признака строго индивидуальный).
Хромосомные и генные мутации оказывают разнообразные воздействия на организм. Во многих случаях эти мутации детальны, так как нару
шают развитие; у человека, например, около 20% беременностей заканчиваются естественным выкидышем в сроки до 12 нед, и в половине таких случаев можно обнаружить хромосомные аномалии. В результате некоторых хромосомных мутаций определенные гены могут оказаться вместе, и их общий эффект может привести к появлению какого-либо «благоприятного» признака. Кроме того, сближение некоторых генов друг с другом делает менее вероятным их разделение в результате крос- синговера, а в случае «благоприятных» генов это создает преимущество. Геномные мутации, наряду с изменением фенотипа, часто приводят к самопроизвольному аборту или хромосомной болезни. Среди новорожденных и детей, умерших в перинатальный период, хромосомные болезни встречаются с частотой 1 случай на 200.
Эпидемиология моногенных (монофакторных) наследственных (менделирующих) заболеваний
Эта группа заболеваний патогенетически и этиологически обусловлена мутациями в одном гене, повреждение которого имеет определяющее значение для эффекта развития болезни. Наследование данного генетического дефекта приводит к повторному появлению, так называемому «выщеплению», патологического фенотипа в пределах конкретной семьи. Результатом таких мутаций обычно является функционально значимый дефект фермента, рецептора, структурного белка клетки или транспортной молекулы. Эти дефекты определяют закономерную причинно-следственную связь между повреждением гена и развитием болезни. Наследование моногенных заболеваний идет в соответствии с законами Менделя, поэтому они называются менделирующими заболеваниями, по имени выдающегося исследователя-генетика Грегора Менделя, сформулировавшего в 1866 г. основные общебиологические законы наследственности (менделевские законы).
Наследование генетических факторов (причин моногенных заболеваний) в пределах родословных семей подчиняется строгим генетическим законам, отражающим характер сегрегации генетического дефекта в ряду поколений. При наследственных менделирующих заболеваниях роль основного генетического локуса в этиопатогенезе болезни является ведущей, однако фенотипическая экспрессия мутации может в определенной степени модифицироваться под действием других факторов как эндогенной, так и экзогенной природы. Например, условий среды проживания, характера питания, особенностей энергетического метаболизма организма и отдельных его органов и тканей, под воздействием разнообразных факторов, в том числе средового характера. Кроме того, модификация экспрессии генов обусловливается действием генов-модификаторов.
Частота распространения
наследственно обусловленных заболеваний
Частота распространения наследственно обусловленных заболеваний зависит от характера наследования генетически измененных признаков и
является достаточно постоянной в той или иной популяции. Однако возможности современной медицины, в том числе, хирургических вмешательств позволяют лицам, имеющим такие дефекты, доживать до возраста половой зрелости и чаще, чем раньше, иметь потомство, что увеличивает частоту индивидуумов с генетическими дефектами в популяции.
Типы наследования моногенных болезней
Тип наследования является важнейшей и постоянной характеристикой любого моногенного заболевания. Он отражает функциональную значимость соответствующего мутантного гена, его хромосомную локализацию и механизмы реализации мутации на клеточном уровне. Менделирующие заболевания могут наследоваться по аутосомно-доминантному, аутосомно-рецессивному и Х-сцепленному типам.
Аутосомно-доминантный тип наследования болезни имеет место в тех случаях, когда патологический ген является доминантным и обеспечивает развитие манифестной формы болезни даже в гетерозиготном состоянии, так как он локализуется на одной из двух гомологичных неполовых хромосом.
Данный тип наследования характеризуется следующими признаками:
• прямая передача болезни происходит от одного из родителей, что является прямой вертикальной передачей генетических признаков, в том числе от больного отца;
• нередко прослеживается манифестация болезни в нескольких поколениях.
Доминантные гены обладают различной пенетрантностью — вероятностью проявления действия мутантного гена у его носителя. При неполной пенетрантности мутантного гена отдельные члены семьи, имеющие мутантный ген, заведомо являющиеся носителями мутации (так называемые «облигатные» носители), могут на протяжении всей жизни оставаться клинически здоровыми, но при этом передать свой мутантный ген потомкам (детям).
Аутосомно-доминантный тип наследования характерен для ряда заболеваний, таких как хорея Гентингтона, нейрофиброматоз, эссенциаль- ный тремор, торсионная дистония, различные формы наследственной дистонии и т. д.
Распространенность хореи Гентингтона в большинстве популяций мира составляет 4—10 случаев на 100 тыс. населения. По сравнению с хореей Гентингтона другие виды наследственных хореических гиперки- незов являются значительно более редкими. Нейрофиброматоз распространен с частотой 28 на 100 тыс. человек. Эссенциальный тремор — наиболее распространенное экстрапирамидальное заболевание человека, случаи заболевания составляют от 0,4 до 6,7% среди лиц моложе 40 лет и достигают 8-13% на восьмом-девятом десятилетии жизни. Другим примером распространенности болезней с аутосомно-доминантным типом наследования является гиперкинетическая форма торсионной дистонии. Известно, что она особенно часто встречается в этнической группе евреев ашкенази — около 40—50 случаев на 100 тыс. Такие формы дистонии, как спастическая кривошея, писчий спазм, спастическая дисфония, встречаются в общей популяции с частотой 3,4 на 100 тыс. населения.
Около 10—15% случаев прионных заболеваний имеют наследственно-семейный характер (семейная форма Крейтцфельдта—Якоба, а также синдром Герстманна—Штреусслера, фатальная семейная инсомния) — они передаются с помощью аутосомно-доминантного типа наследования. Развитие семейных форм прионных болезней связано с наследованием мутаций в гене прионного белка РRNР, который локализован в дистальном участке короткого плеча 20-й хромосомы, точная функция его до сих пор не установлена.
Структура заболеваемости имеет следующие особенности:
• соотношение больных и здоровых лиц у потомков больного индивидуума близко к 50%, соответственно, для каждого из детей — потомков больного родителя риск унаследовать мутантный ген, т. е. риск возникновения заболевания, равен 50%;
• половая структура заболевших представлена поровну мужчинами и женщинами, так как оба пола поражаются в равной степени, в редких случаях может наблюдаться более высокая пенетрантность и, следовательно, более тяжелое течение заболевания у определенного пола (чаще у женщин).
Аутосомно-рецессивный тип наследования реализуется при наследственной передаче заболеваний, для клинической манифестации которых необходимо присутствие мутантного гена в гомозиготном состоянии, т. е. на обеих гомологичных хромосомах, унаследованных от родителей. Чаще всего при аутосомно-рецессивных заболеваниях первичный молекулярный дефект заключается в повреждении фермента, а патологический эффект проявляется при критическом снижении его активности — ниже порогового значения. Гетерозиготные носители имеют «одинарную дозу» мутации, благодаря второй (нормальной) копии гена активность белка у них составляет около 50%, что обычно вполне достаточно для поддержания соответствующей функции на физиологическом уровне, поэтому эти индивидуумы остаются клинически здоровыми.
Данный тип наследования характеризуется следующими признаками:
• болезнь проявляется в одном поколении среди сибсов (т. е. среди братьев, сестер — детей одной родительской пары), родители при этом остаются здоровыми;
• у родителей больных лиц часто имеет место кровнородственный брак (именно в таком браке наиболее высока вероятность того, что ребенок унаследует от обоих родителей две мутантные хромосомы, имеющие общее генетическое происхождение).
Распространенность. Примером патологии, передающейся с помощью аутосомно-рецессивного типа, наследования, является ювенильный паркинсонизм, распространенность его повсеместная. Другим примером служит болезнь Фридрейха — наиболее часто встречающаяся форма наследственной атаксии. В европейских популяциях распространенность заболевания составляет 2-5 случаев на 100 тыс. человек, а частота гетеро- зиготного носительства мутации около 1 случая на 100 тыс. Частота встречаемости другой формы наследственной атаксии — синдрома Луи- Бар, или синдрома Бодера-Седжвика, составляет 1 на 100 тыс. Еще одним примером заболевания, передающегося через аутосомно-рецессив- ный тип, является миоклонус-эпилепсия Унферрихта—Лундборга, его распространенность составляет в среднем 1 случай на 100 тыс. человек, но в некоторых популяциях — в Финляндии, Северной Африке — гораздо выше — 5 на 100 тыс. Миотоническая дистрофия представляет собой наиболее частую форму мышечной дистрофии у взрослых, передается она также с помощью аутосомно-рецессивного типа наследования и имеет частоту 13 на 100 тыс. населения.
Структура заболеваемости с аутосомно-рецессивным типом наследования имеет следующие особенности:
• доля пораженных сибсов среди всех потомков родительской пары составляет около 25%, риск развития заболевания у каждого ребенка также составляет 25%;
• оба пола поражаются в равной степени.
Х-сцепленный тип наследования. При локализации мутантного гена в Х-хромосоме имеет место наследование, сцепленное с полом. В абсолютном большинстве случаев такие гены являются рецессивными, а тип наследования определяется как Х-сцепленный рецессивный. Поскольку X и У хромосомы не комплементарны, у мужчин даже рецессивный ген, расположенный на единственной Х-хромосоме, не имеет своей пары (состояние гемизиготности) и является манифестирующим. У женщин-гетерозигот мутация на одной из Х-хромосом компенсируется нормальным геном, расположенным на второй копии Х-хромосомы. Таким образом, при Х-сцепленном рецессивном типе наследования заболевание проявляется у мужчин, унаследовавших от матери мутантную хромосому.
Распространенность заболеваний с Х-сцепленным типом наследования может быть продемонстрирована на примере прогрессирующих мышечных дистрофий, таких как дистрофии Дюшенна и Бекера, которые являются самыми распространенными среди мышечных дистрофий и имеют частоту 28 на 100 тыс. и 5 на 100 тыс. соответственно.
Данный тип наследования характеризуется следующими признаками:
• заболевание проявляется только у мужчин;
• заболевание передается следующему поколению (половине своих сыновей) здоровыми клинически женщинами-носителями — через передачу им мутантной Х-хромосомы;
• отсутствует прямая передача болезни от мужчин их сыновьям, поскольку сыновья всегда наследуют от отца нормальную У-хро- мосому;
• все дочери больных мужчин являются клинически здоровыми гетерозиготными носителями мутировавшего гена.
Х-сцепленный доминантный тип наследования. Этот тип наследования формируется, когда ген, локализованный на Х-хромосоме, определяет развитие доминантного признака.
Для этого типа наследования генетических признаков характерны следующие особенности;
• все дочери больного отца наследуют заболевание;
• передача от отца сыну невозможна, так как сыновья наследуют от отца здоровую У-хромосому,
• вероятность рождения больного ребенка любого пола от больной матери равна 50%;
• в каждой родословной число больных женщин в 2 раза больше, чем больных мужчин
Группы риска заболеваний
моногенными формами патологии
Группами риска при моногенных болезнях являются, как правило, следующие категории:
• дети, у которых родители, сибсы или другие родственники имеют наследственное заболевание;
• лица, которые в связи с близким родством с пробандом имеют повышенный риск гетерозиготного носительства мутантного гена (гетерозиготами являются родители и дети гомозигот по рецессивному типу);
• при болезнях с доминантным типом наследования с неполной пенетрантностью носителями патологического гена являются все лица, имеющие больных детей и больных родителей одновременно;
• при сцепленных с Х-хромосомой болезнях гетерозиготными «кондукторами» являются все дочери больного и все матери больных (гемофилия).
Примерами принадлежности к группам риска при наличии хромосомной патологии являются следующие:
• дети матерей в возрасте старше 36 лет (риск рождения ребенка с синдромом Дауна в 40 раз больше, чем у 20-летней женщины);
• при наличии в семье детей с хромосомными болезнями,
• отягощенный акушерский анамнез матери и семейный анамнез (выкидыши, мертворождения, дети с множественными врожденными пороками развития, дети погибшие с неустановленным диагнозом, особенно если у матери есть микроаномалии и единичные врожденные пороки развития, которые могут быть признаком мозаицизма при хромосомной аберрации);
• наличие у матери (отца) хромосомного мозаицизма или хромосомной аберрации, установленной ранее;
• контакт родителей с мутагенными факторами.
Профилактика моногенных наследственных заболеваний.
Выявление ведущих факторов риска
Только при использовании аналитических эпидемиологических методов исследования возможно выявление ведущих факторов риска. Это демонстрирует пример изучения факторов риска рождения детей с синдромом Дауна, ставший хрестоматийным Частота рождения детей с синдромом Дауна возрастает в зависимости от очередности рождения ребенка, так, этот показатель увеличивается почти в три раза для детей, родившимися в семье пятыми и более (рис 34 1).
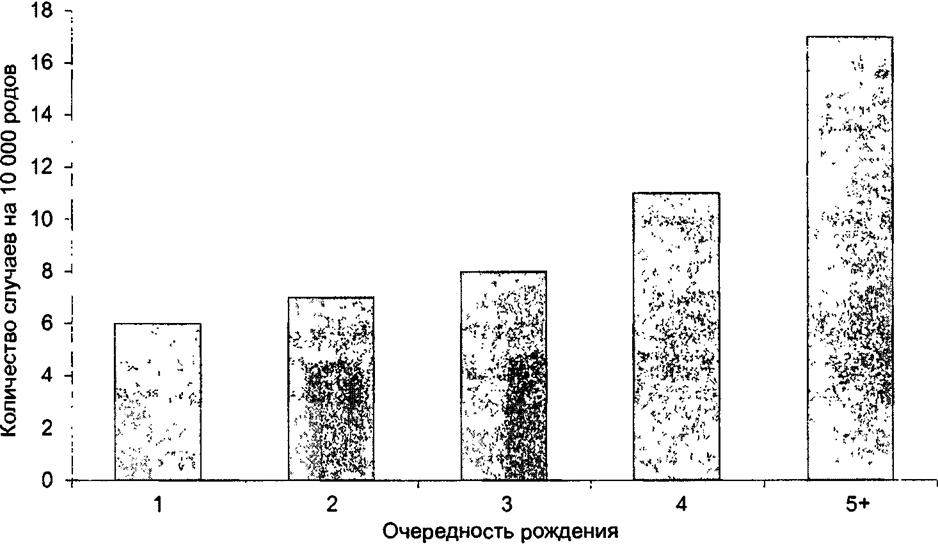
Рис. 34.1. Частота возникновения синдрома Дауна в зависимости от очередности рождения |
Однако еще более выраженная зависимость (возрастание риска почти в 40 раз) существует между частотой рождения детей с болезнью Дауна и возрастом матери (рис 34.2). Очевидно, что обе переменные (очередность рождения и возраст матери) коррелируют: женщина, рожающая пятого ребенка, более вероятно окажется старше матери первого или второго ребенка Поэтому справедливым будет предположить, что фактор очередности рождения искажает влияние возраста матери Для того чтобы понять, так ли это, разумно изучить влияние обоих факторов одновременно (рис 34.3). Из рисунка хорошо видно, что для каждой категории по порядку рождения риск рождения ребенка с синдромом Дауна увеличивается с возрастанием возраста матери. В то же время внутри каждой возрастной категории не наблюдается никакого влияния очередности рождения* это означает, что кажущийся эффект зависимости развития синдрома Дауна от очередности рождения ребенка действительно является результатом искажения.
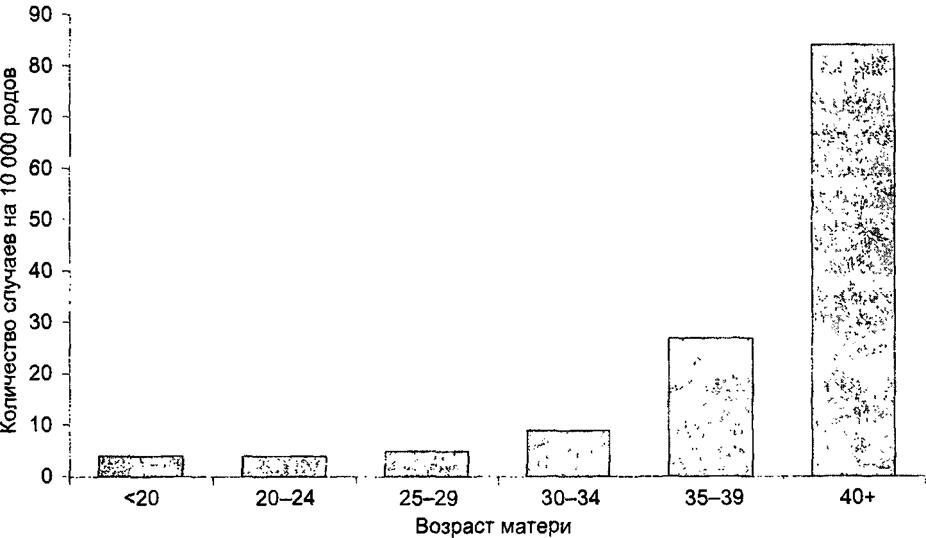
Рис. 34.2. Частота возникновения синдрома Дауна в зависимости от возраста матери |
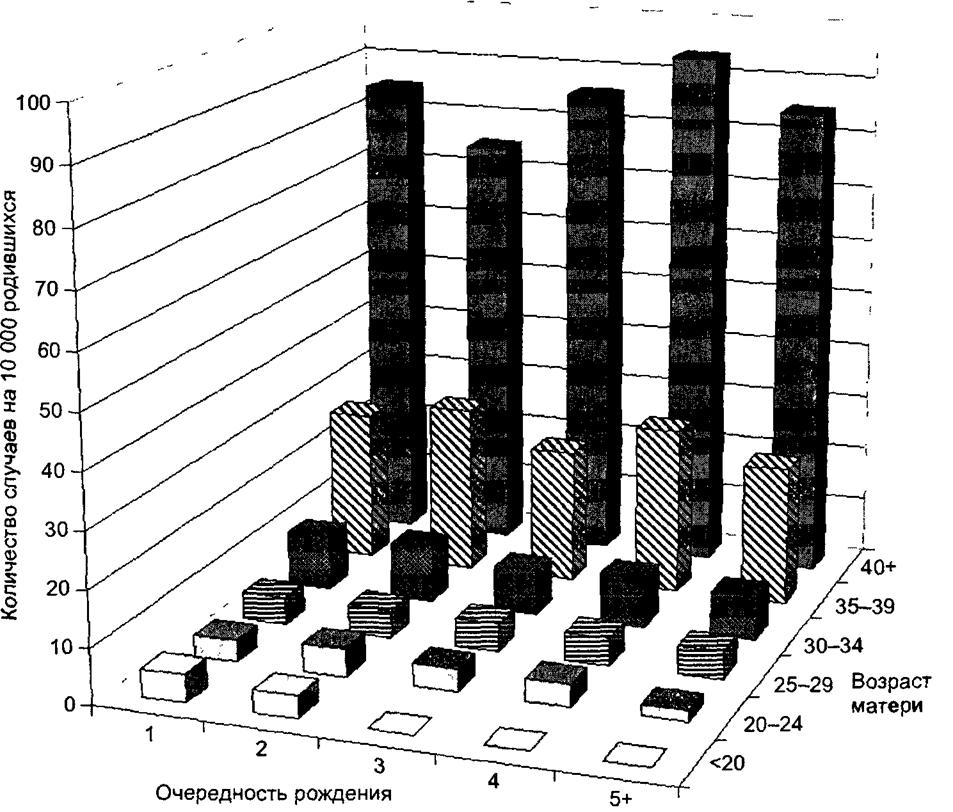
Рис. 34.3. Частота возникновения синдрома Дауна в зависимости от очередности рождения и возраста матери |
В геноме человека насчитывается 35—50 тыс. различных генов, изменения в некоторых из них приводят к нескольким тысячам наследственных болезней. Гены работают в весьма тесной зависимости друг от друга — информация «распределена» между ними. Кроме того, такое небольшое количество генов означает, что в биологии человека очень многое зависит от характера окружающей среды. Гены практически всех наиболее частых (около 320) и сравнительно редких (около 170) наследственных болезней уже известны. Методы их обнаружения достаточно просты и универсальны и поэтому широко применяются в медицине. Выявление генов наследственных болезней на ранних сроках беременности (с десятой недели) позволяет предотвратить рождение больного ребенка. Впервые в нашей стране внутриутробный диагноз (гемофилии) был поставлен в 1989 г. в Санкт-Петербурге в Институте акушерства и гинекологии имени Д. О. Отта. Затем здесь же впервые в России пренатальная диагностика позволила выявить также такие тяжелейшие генные патологии, как муковисцидоз, миодистрофия Дюшенна, фенилкетонурия, синдром ломкой Х-хромосомы. Прекращение беременности — хотя и эффективный, но достаточно грубый способ охраны генофонда и здоровья живущего поколения. Поэтому значительно более привлекательным представляется такое направление профилактики, как выявление бессимптомных взрослых носителей того или иного наследственного заболевания. Профилактическим мероприятием в данном случае будет предупреждение человека о вероятности рождения у него больного ребенка и оценка степени риска.
Источник: Л. П. Зуева, Р. X. Яфаев, «ЭПИДЕМИОЛОГИЯ» 2005