Аномалии развития вентрикулярной системы и подпаутинного пространства
Пороки развития вентрикулярной системы обычно возникают в области ее анатомических сужений: межжелудочковых отверстий, водопровода среднего мозга, срединной и латеральной апертур IV желудочка. Это — главным образом стенозы и атрезии названных сужений, приводящие к развитию внутренней водянки головного мозга.
Атрезия водопровода мозга в толще ножек мозга обнаруживаются мелкие, слепо заканчивающиеся трубчатые ходы из клеток эпендимы, расположенные беспорядочно по всему веществу ножек.
Атрезия межжелудочковых отверстий (син.: атрезия отверстии Монро) может быть результатом неправильного развития или перенесенного воспалительного процесса, встречается редко. При сужении одного из отверстий Монро развивается асимметричная гидроцефалия.
Атрезия отверстия Мажанди (atresia foraminis Afagandie)- атрезия срединного отверстия IV желудочка, сопровождается расстройством циркуляции спинно-мозговой жидкости (внутренняя гидроцефалия), в отдельных случаях протекает бессимптомно.
Атрезия срединной и латеральных апертур IV желудочка — обычно сопровождается развитием синдрома (порока) Денди — Уокера. Нередко этот порок сочетается с другими аномалиями головного мозга (микрогирией. полигирией или пахигирией. агенезией мозолистого тела, гетеротопиями клеток коры в белое вещество) (рис. 9).
Гидранэнцефалия полное или почти полное отсутствие больших полушарий при сохранности костей свода черепа и мягких покровов головы. Голова при этом пороке нормальных размеров или несколько увеличена. Полость черепа заполнена прозрачной спинномозговой жидкостью. Продолговатый мозг и мозжечок сохранены. Крайне редкий порок.
Гидроцефалия врожденная водянка головного мозга, чрезмерное накопление в вентрикулярной системе или подпаутинном пространстве спинно-мозговой жидкости, сопровождающееся атрофией мозгового вещества. Большинство случаев врожденной гидроцефалии обусловлено нарушениями оттока спинно-мозговой жидкости в подпаутинное пространство. Нарушение оттока может быть вызвано стенозом или атрезией сильвиева водопровода, атрезией отверстий Люшки и Мажанди, аномалиями основания черепа. Атрезия отверстий Лушки и Мажанди сопровождается пороком Денди - Уокера. Намного реже врожденная гидроцефалия может явиться следствием повышенной продукции спинномозговой жидкости (гидроцефалия гиперсекреторная) или ее пониженной резорбции (гидроцефалия арезорбтивная). Клинико-морфологически выделяют 2 вида:
а) гидроцефалия внутренняя (син.: гидроцефалия закрытая, гидроцефалия окклюзионная) — спинно-мозговая жидкость накапливается в вентрикулярной системе;
б) гидроцефалия наружная (син.: гидроцефалия открытая, гидроцефалия сообщающаяся) - спинно-мозговая жидкость накапливается в подпаутинном пространстве (рис. 10).
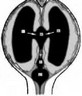
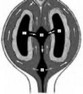 Рис. 9. Схема патологических изменений при синдро- Рис. 10. Схема цир^ляции спинно-мозговой жпдкс- ме Денди-Уокера (Romero R., Pilu D., Genty F., 1997): сти при сообщающейся гндроцефашш, развивающейся четвертый желудочек (IV) сообщается с kuci ой задней в результате блокады реабсорбции спинно-мозговой черепной ямки (CV). Блокада оттока спинно-мозговой жидкости ив уровне верхнего carni юльного синуса жидкости ив уровне отверстий Люшки и Мажанди (X) (lt;^7). (Romero R., Pilu D., Genty F., 1997): скопление приводи! к увеличению IV, III и боковых (I и П) желу- спинно-мозговой жидкости приводит к одновремен- дочков. верхний сатпиальный синус ному расширению желудочков и субарахноидального
Рис. 9. Схема патологических изменений при синдро- Рис. 10. Схема цир^ляции спинно-мозговой жпдкс- ме Денди-Уокера (Romero R., Pilu D., Genty F., 1997): сти при сообщающейся гндроцефашш, развивающейся четвертый желудочек (IV) сообщается с kuci ой задней в результате блокады реабсорбции спинно-мозговой черепной ямки (CV). Блокада оттока спинно-мозговой жидкости ив уровне верхнего carni юльного синуса жидкости ив уровне отверстий Люшки и Мажанди (X) (lt;^7). (Romero R., Pilu D., Genty F., 1997): скопление приводи! к увеличению IV, III и боковых (I и П) желу- спинно-мозговой жидкости приводит к одновремен- дочков. верхний сатпиальный синус ному расширению желудочков и субарахноидального
пространства I, П — боковые желудочки, III — 3-й желудочек, IV — 4-й желудочек, заштрихованная часш - су барах но ид альное пространство
Обеим формам свойственны общие признаки. Увеличение окрз'жности головы до 50-70 см (при норме 34-35 см). В 30° о случаев увеличение объема головы наблюдается при рождении, в 50° о — через 3 месяца после рождения. Отмечаются истончение и расхождение костей черепа, выраженная подкожная венозная сеть, выбз'хание родничков, диспропорция мозговой и лицевой частей черепа — маленькое лицо, нависающий лоб. Волосы на голове редкие. Повьппение внутричерепного давления сопровождается неврологической симптоматикой: рвотой, косоглазием, спастическими парезами с повышением сз'хожильных рефлексов, нарзчлением координации. Характерна задержка умственного развития. На глазном дне отмечаются застойные явления и отек соска зрительного нерва В слз'чае деформации костных стрз'кгз'р основания черепа могут возникизть симптомы сдавления мозжечка, ствола мозга и верхней части спинного мозга, патология со стороны черепных нервов, расстройства движений и координации, ннстагм. Популяционная частота- 1:2000.
Дивертикул водопровода мозга — слепое мешкообразное выпячивание стенки водопровода мозга, сопровождается гидроцефалией. Может быть одиночным или множественным. Облитерация субарахноидального пространства врожденная — отсзтствие подпазтинного пространства головного мозга вследствие сращения мягкой и пазтинной оболочек, встречается крайне редко.
Расщепление водопровода мозга — представляет собой разделение на два какала: основной дорсальный и меньший — вентральный. Иногда впереди от основного какала располагается большое количество мелких трз'бчатых ходов, построенных из эпендимального эпителия. Главный и добавочный каналы разделены неизмененной нервной тканью.
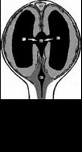
Стеноз водопровода мозга - врожденное сужение водопровода мозга; отмечаются пщроцефашля. уве личенне объема черепа, расхождение черепных швов; задержка умственного н физического развития; симптомы выпадения функций черепных нервов В ряде случаев сопровождается гпиозом периакведукталъной зоны. Рецессивное, сцепленное с Х-хромосомой наследование (рнс. II).
 Комбинированные пороки развития
Комбинированные пороки развития
Арнольда — Кнарн синдром (Arnold— Chiari синдром, син.: morbus Arnold — Chiari, anomalia Arnold — Chiari, dysraphia cerebelli) - обусловлен пороком развития ствола мозга, прн котором имеется кау дальное смещение продолговатого мозга, моста, червя мозжечка н удлинение полостн IV желудочка. Червь мозжечка, продолговатый мозг н IV желудочек располагаются в верхнешейиой части позвоночного канала. Почтн всегда сочетается с миеломенпнгоцеле. Порок обусловлен асинхронным ростом ствола мозга н спинного мозга. Клинически - мозжечковые расстройства с атаксией н ннстагмом; признаки сдавления ствола мозга н спинного мозга - параличи черепных нервов, приступы тетаноидных или эпилептнформных судорог, диплопия, гемиа- нопсия. Часто сочетается со стенозом водопровода, мзжрошрнеп, недоразвшнем четверохолмия, платибазией, спмподиеи, аномалиями черепа н шейных позвонков. Аугосомно- рецессивное наследование.
Бикерса — Адамса синдром (Bickers—Adams синдром, син.. стеноз водопровода мозга) — наследственный стеноз водопровода мозга; отмечаются увеличение объема черепа, расхождение черепных швов; задержка умственного н физического развития; симптомы выпадения функций черепных нервов, спастическая параплешя; гипоплазия и контрактуры больших пальцев рук. В ряде случаев сопровождается глиозом периакведукталъной зоны. Рецессивное, сцепленное с Х-хромосомой наследование.
Денди — Уокера синдром (Dandy — Walker синдром, с-ин.: Денди — Уокера порак, atresia forammis Alagandie) - врожденная аномалия мозга в области IV желудочка с расстройством циркупящш спинно-мозговой жидкости, характеризуется триадой: внутренняя гидроцефалия, гипоплазия или аплазия червя мозжечка, кистозное расширение IV желудочка. Возникает при атрезии срединного отверстия IV желудочка (в отдельных случаях протекает бессимптомно). Аугосомно-рецессивное наследование
Кундрата синдром (Кипdrat синдром, син.: atrhinencephajia) - аплазия обонятельных луковиц, борозд, трактов н пластинок, с нарушением в ряде случаев гиппокампа. Сопровождается аплазией продырявленной пластинки решетчатой костн н петушиного гребня, отсутствием или гипоплазией прямых извилин лобных долей, агенезией костей носа, гипотело- ризмом (иногда - циклопией) н другими пороками развития черепа. Аугосомно- рецессивное наследование.
Миллера — Дикера синдром (сии.: лиссэнцефстия, агирия) — наиболее важный диагностический признак микроцефалия (100° о). Типичен внешний вид больных: высокий лоб, суженный в височных областях, выступающий затылок, ротированные назад ушные раковины со сглаженным рисунком, антимонголоидный разрез глаз, гипертелоризм, микро- гнатия, «рыбий рот», усиленное оволосение лица. Отмечаются помутнение роговицы, полидактилия, камптодактилия, неполная кожная синдактилия II III пальцев стоп, поперечная ладонная складка, морщинистая кожа. Кроме того, описаны врожденные пороки сердца, агенезия почек, атрезия двенадцатиперстной кишки, крипторхизм, паховые грыжи. Характерны мышечная гипотония, затруднение глотания, эпизоды апноэ с цианозом, повышение сухожильных рефлексов, опистотонус и децеребрационная ригидность, судорожные приступы с l-й недели жизни, выраженное отставание в психомоторном развитии. Имеются неспецифические изменения на пневмоэнцефалограммах. Больные погибают в раннем детстве. На аутопсии обнаруживают отсутствие борозд и извилин в больших полушариях головного мозга, недоразвитие серого вещества, возможны также расширение IV желудочка и гипоплазия средних отделов мозжечка. Тип наследования — аутосом- но-рецессивный.
Атрезия водопровода мозга в толще ножек мозга обнаруживаются мелкие, слепо заканчивающиеся трубчатые ходы из клеток эпендимы, расположенные беспорядочно по всему веществу ножек.
Атрезия межжелудочковых отверстий (син.: атрезия отверстии Монро) может быть результатом неправильного развития или перенесенного воспалительного процесса, встречается редко. При сужении одного из отверстий Монро развивается асимметричная гидроцефалия.
Атрезия отверстия Мажанди (atresia foraminis Afagandie)- атрезия срединного отверстия IV желудочка, сопровождается расстройством циркуляции спинно-мозговой жидкости (внутренняя гидроцефалия), в отдельных случаях протекает бессимптомно.
Атрезия срединной и латеральных апертур IV желудочка — обычно сопровождается развитием синдрома (порока) Денди — Уокера. Нередко этот порок сочетается с другими аномалиями головного мозга (микрогирией. полигирией или пахигирией. агенезией мозолистого тела, гетеротопиями клеток коры в белое вещество) (рис. 9).
Гидранэнцефалия полное или почти полное отсутствие больших полушарий при сохранности костей свода черепа и мягких покровов головы. Голова при этом пороке нормальных размеров или несколько увеличена. Полость черепа заполнена прозрачной спинномозговой жидкостью. Продолговатый мозг и мозжечок сохранены. Крайне редкий порок.
Гидроцефалия врожденная водянка головного мозга, чрезмерное накопление в вентрикулярной системе или подпаутинном пространстве спинно-мозговой жидкости, сопровождающееся атрофией мозгового вещества. Большинство случаев врожденной гидроцефалии обусловлено нарушениями оттока спинно-мозговой жидкости в подпаутинное пространство. Нарушение оттока может быть вызвано стенозом или атрезией сильвиева водопровода, атрезией отверстий Люшки и Мажанди, аномалиями основания черепа. Атрезия отверстий Лушки и Мажанди сопровождается пороком Денди - Уокера. Намного реже врожденная гидроцефалия может явиться следствием повышенной продукции спинномозговой жидкости (гидроцефалия гиперсекреторная) или ее пониженной резорбции (гидроцефалия арезорбтивная). Клинико-морфологически выделяют 2 вида:
а) гидроцефалия внутренняя (син.: гидроцефалия закрытая, гидроцефалия окклюзионная) — спинно-мозговая жидкость накапливается в вентрикулярной системе;
б) гидроцефалия наружная (син.: гидроцефалия открытая, гидроцефалия сообщающаяся) - спинно-мозговая жидкость накапливается в подпаутинном пространстве (рис. 10).
Рис. 9. Схема патологических изменений при синдро- Рис. 10. Схема цир^ляции спинно-мозговой жпдкс- ме Денди-Уокера (Romero R., Pilu D., Genty F., 1997): сти при сообщающейся гндроцефашш, развивающейся четвертый желудочек (IV) сообщается с kuci ой задней в результате блокады реабсорбции спинно-мозговой черепной ямки (CV). Блокада оттока спинно-мозговой жидкости ив уровне верхнего carni юльного синуса жидкости ив уровне отверстий Люшки и Мажанди (X) (lt;^7). (Romero R., Pilu D., Genty F., 1997): скопление приводи! к увеличению IV, III и боковых (I и П) желу- спинно-мозговой жидкости приводит к одновремен- дочков. верхний сатпиальный синус ному расширению желудочков и субарахноидального
пространства I, П — боковые желудочки, III — 3-й желудочек, IV — 4-й желудочек, заштрихованная часш - су барах но ид альное пространство
Обеим формам свойственны общие признаки. Увеличение окрз'жности головы до 50-70 см (при норме 34-35 см). В 30° о случаев увеличение объема головы наблюдается при рождении, в 50° о — через 3 месяца после рождения. Отмечаются истончение и расхождение костей черепа, выраженная подкожная венозная сеть, выбз'хание родничков, диспропорция мозговой и лицевой частей черепа — маленькое лицо, нависающий лоб. Волосы на голове редкие. Повьппение внутричерепного давления сопровождается неврологической симптоматикой: рвотой, косоглазием, спастическими парезами с повышением сз'хожильных рефлексов, нарзчлением координации. Характерна задержка умственного развития. На глазном дне отмечаются застойные явления и отек соска зрительного нерва В слз'чае деформации костных стрз'кгз'р основания черепа могут возникизть симптомы сдавления мозжечка, ствола мозга и верхней части спинного мозга, патология со стороны черепных нервов, расстройства движений и координации, ннстагм. Популяционная частота- 1:2000.
Дивертикул водопровода мозга — слепое мешкообразное выпячивание стенки водопровода мозга, сопровождается гидроцефалией. Может быть одиночным или множественным. Облитерация субарахноидального пространства врожденная — отсзтствие подпазтинного пространства головного мозга вследствие сращения мягкой и пазтинной оболочек, встречается крайне редко.
Расщепление водопровода мозга — представляет собой разделение на два какала: основной дорсальный и меньший — вентральный. Иногда впереди от основного какала располагается большое количество мелких трз'бчатых ходов, построенных из эпендимального эпителия. Главный и добавочный каналы разделены неизмененной нервной тканью.
Стеноз водопровода мозга - врожденное сужение водопровода мозга; отмечаются пщроцефашля. уве личенне объема черепа, расхождение черепных швов; задержка умственного н физического развития; симптомы выпадения функций черепных нервов В ряде случаев сопровождается гпиозом периакведукталъной зоны. Рецессивное, сцепленное с Х-хромосомой наследование (рнс. II).
Комбинированные пороки развития
Арнольда — Кнарн синдром (Arnold— Chiari синдром, син.: morbus Arnold — Chiari, anomalia Arnold — Chiari, dysraphia cerebelli) - обусловлен пороком развития ствола мозга, прн котором имеется кау дальное смещение продолговатого мозга, моста, червя мозжечка н удлинение полостн IV желудочка. Червь мозжечка, продолговатый мозг н IV желудочек располагаются в верхнешейиой части позвоночного канала. Почтн всегда сочетается с миеломенпнгоцеле. Порок обусловлен асинхронным ростом ствола мозга н спинного мозга. Клинически - мозжечковые расстройства с атаксией н ннстагмом; признаки сдавления ствола мозга н спинного мозга - параличи черепных нервов, приступы тетаноидных или эпилептнформных судорог, диплопия, гемиа- нопсия. Часто сочетается со стенозом водопровода, мзжрошрнеп, недоразвшнем четверохолмия, платибазией, спмподиеи, аномалиями черепа н шейных позвонков. Аугосомно- рецессивное наследование.
Бикерса — Адамса синдром (Bickers—Adams синдром, син.. стеноз водопровода мозга) — наследственный стеноз водопровода мозга; отмечаются увеличение объема черепа, расхождение черепных швов; задержка умственного н физического развития; симптомы выпадения функций черепных нервов, спастическая параплешя; гипоплазия и контрактуры больших пальцев рук. В ряде случаев сопровождается глиозом периакведукталъной зоны. Рецессивное, сцепленное с Х-хромосомой наследование.
Денди — Уокера синдром (Dandy — Walker синдром, с-ин.: Денди — Уокера порак, atresia forammis Alagandie) - врожденная аномалия мозга в области IV желудочка с расстройством циркупящш спинно-мозговой жидкости, характеризуется триадой: внутренняя гидроцефалия, гипоплазия или аплазия червя мозжечка, кистозное расширение IV желудочка. Возникает при атрезии срединного отверстия IV желудочка (в отдельных случаях протекает бессимптомно). Аугосомно-рецессивное наследование
Кундрата синдром (Кипdrat синдром, син.: atrhinencephajia) - аплазия обонятельных луковиц, борозд, трактов н пластинок, с нарушением в ряде случаев гиппокампа. Сопровождается аплазией продырявленной пластинки решетчатой костн н петушиного гребня, отсутствием или гипоплазией прямых извилин лобных долей, агенезией костей носа, гипотело- ризмом (иногда - циклопией) н другими пороками развития черепа. Аугосомно- рецессивное наследование.
Миллера — Дикера синдром (сии.: лиссэнцефстия, агирия) — наиболее важный диагностический признак микроцефалия (100° о). Типичен внешний вид больных: высокий лоб, суженный в височных областях, выступающий затылок, ротированные назад ушные раковины со сглаженным рисунком, антимонголоидный разрез глаз, гипертелоризм, микро- гнатия, «рыбий рот», усиленное оволосение лица. Отмечаются помутнение роговицы, полидактилия, камптодактилия, неполная кожная синдактилия II III пальцев стоп, поперечная ладонная складка, морщинистая кожа. Кроме того, описаны врожденные пороки сердца, агенезия почек, атрезия двенадцатиперстной кишки, крипторхизм, паховые грыжи. Характерны мышечная гипотония, затруднение глотания, эпизоды апноэ с цианозом, повышение сухожильных рефлексов, опистотонус и децеребрационная ригидность, судорожные приступы с l-й недели жизни, выраженное отставание в психомоторном развитии. Имеются неспецифические изменения на пневмоэнцефалограммах. Больные погибают в раннем детстве. На аутопсии обнаруживают отсутствие борозд и извилин в больших полушариях головного мозга, недоразвитие серого вещества, возможны также расширение IV желудочка и гипоплазия средних отделов мозжечка. Тип наследования — аутосом- но-рецессивный.
Источник: Калмин О.В. , «Аномалии развития органов и частей тела человека» 2004