ГИПЕРГЛИКЕМИЯ, ИНСУЛИНОРЕЗИСТЕНТНОСТЬ И ОКСИДАТИВНЫЙ СТРЕСС
ГИПЕРГЛИКЕМИЯ
Гипергликемия характерна как для диабета типа 1, так и для диабета типа 2. В нескольких исследованиях была выявлена связь между повышением концентрации глюкозы в плазме крови и возрастанием сердечно-сосудистой смертности и заболеваемости [14]. Растущие усилия направлены на выяснение влияния глюкозы на функцию сосудов, в том числе на эндотелиальную функцию и биодоступность оксида азота (NO).
Эндотелий вносит вклад в контроль тонуса гладкой мускулатуры стенки сосудов путем выделения NO, вызывающего вазодилатацию и ингибирующего тромбоциты (т.е. предотвращающего сосудистый спазм и формирование тромба).
Реакцию образования NO из терминального гуанидинового атома азота L-аргинина катализируют ферменты семейства NO-синтаз (NOSs). Один из этих ферментов, эндотелиальная NO-синтаза (eNOS), Ca2+-зависима. Она постоянно находится в клетках различных типов, включая клетки эндотелия.
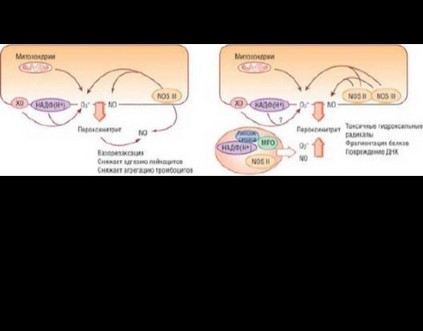
Активность превращения L-аргинина в NO определяется балансом между синтезом и распадом NO путем его реакции с супероксид-анионом (O2-). В физиологических условиях O2- заметно не влияет на образование этой молекулы, поэтому NO может оказывать свое протективное действие на сосуды, способствуя поддержанию антиатеросклеротических условий. Однако при наличии факторов сердечно-сосудистого риска продукция супероксид-аниона быстро становится избыточной, NO инактивируется, повышается концентрация пероксинитрита (ONOO-) - очень мощного оксиданта (рис. 14.3).
 итзидрргы j лт;р«иgt;еии |
итзидрргы j лт;р«иgt;еии |
образования супероксид-аниона [15-17] (рис. 14.4). Потеря биодоступности оксида азота предшествует развитию необратимого атеросклероза и служит независимым предиктором неблагоприятных сердечно-сосудистых событий [18].
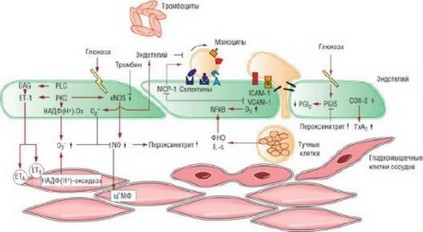
Рис. 14.4. Гипергликемия и производимые эндотелием вазоактивные субстанции. ФНО - фактор некроза опухолей; НАДФ(Н+) - восстановленная форма
никотинамидадениндинуклеотидфосфата; цГМФ - циклический гуанозинмонофосфат; СОХ- 2 - циклооксигеназа-2; DAG - диацилглицерин; eNOS - эндотелиальная NO-синтаза; ЕТ - эндотелин;^-в - интерлейкины; МСР-1 - моноцитарный белок-хемоаттрактант-1; NFkB - нуклеарный фактор kB; PGIS - простациклинсинтаза; PGI2 - простациклин; РКС - протеинкиназа С; PLC - фосфолипаза С; ТхА2 - тромбоксан А2; VCAM-1 - сосудистые молекулы клеточной адгезии. Источник (с разрешения): Creager M.A., Luscher T.F.,
Cosentino F. et al. Diabetes and vascular disease: pathophysiology, clinical consequences and medical therapy: Part I // Circulation. - 2003. - Vol. 108. - P. 1527-1532.
Митохондриальная продукция O2- - важный медиатор гипергликемического поражения сосудов [16]. Дальнейшее повышение концентрации O2- происходит под действием порочного круга, в котором свободные кислородные радикалы (ROS - от reactive oxygen species, частицы активного кислорода) индуцируют активацию протеинкиназы C [15] и приводят к повышению протеинкиназа C-опосредованной продукции ROS. Показано, что активация протеинкиназы С глюкозой вовлечена в регуляцию и активацию НАД(Ф)^-зависимой оксидазы - важного сосудистого источника продукции O2- [16, 19]. Активность НАД(Ф)H+ и экспрессия субъединиц белка у пациентов с диабетом повышены во внутренних грудных артериях и больших подкожных венах бедра [20]. Хотя активация протеинкиназы С под действием высокой концентрации глюкозы ведет к ''up''-регуляции (т.е. к позитивной регуляции) экспрессии NO-синтазы, повышенный распад оксида азота перекрывает этот эффект и вызывает в конечном счете сокращение биодоступности NO. Таким образом, активация протеинкиназного пути - тот узел во внутриклеточной передаче сигнала, который ведет к индуцированному гипергликемией оксидативному стрессу и эндотелиальной дисфункции [21] (рис. 14.5).
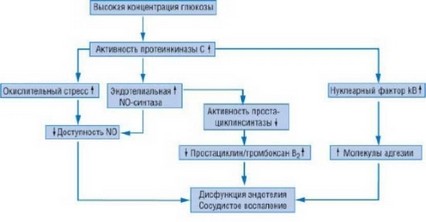
Рис. 14.5. Объединенный протеинкиназа C-зависимый механизм служит триггером, при помощи которого гипергликемия индуцирует эндотелиальную дисфункцию и сосудистое воспаление. NO - оксид азота.
Избыток свободных кислородных радикалов влияет на эндотелиальную функцию несколькими путями.
Супероксид-анион быстро инактивирует NO и превращает его в пероксинитрит [22] - мощный оксидант, который легко проходит через фосфолипидные мембраны и вызывает нитрование субстратов, блокируя регуляторные рецепторы, ферменты-инактиваторы свободных радикалов [23, 24] и ключевые кофакторы эндотелиальной NO-синтазы (eNOS), например тетрагидробиоптерин [25].
Митохондриальная продукция супероксид-аниона повышает внутриклеточное образование продуктов конечного гликози-лирования, что неблагоприятно отра-жается на эндотелиальной функции в связи с усилением образования свободных радикалов и провоспалительных цитокинов в клетках сосудов и увеличением эндотелиальной экспрессии различных молекул адгезии, участвующих в атерогенезе [26].
Активация рецепторов продуктов конечного гликозилирования повышает внутриклеточное образование супероксид-аниона [27] и, вероятно, представляет собой ключевую ступень в развитии атеросклеротического поражения [28].
Продукция супероксид-аниона активирует гексозаминный путь, который снижает активацию NOS под действием протеинкиназы Akt [29]. Активация Akt далее ограничивается протеинкиназа С- зависимым ингибированием фосфатидилинозитол-3-киназного пути.
Оксидативный стресс в результате повышения концентрации глюкозы увеличивает содержание диметиларгинина - конкурентного антагониста NOS [30].
Белок-адаптор p66Shc контролирует клеточный ответ на оксидативный стресс. Для лабораторных крыс с дефицитом белка p66Shc(p66Shc-/-) характерна повышенная резистентность к свободным кислородным радикалам (ROS), большая продолжительность жизни и менее выраженный атеросклеротический процесс при высоком потреблении жиров. Это свидетельствует о положительном влиянии p66Shc на процесс старения и появление заболеваний с возрастом [31, 32]. Показано также, что у старых крыс с дефицитом белка p66Shc сохраняется, по сравнению с дикими сородичами того же возраста из того же помета, способность к эндотелийзависимой релаксации сосудов [33]. Кроме того, в клетках с фенотипом p66Shc-/- снижена внутриклеточная концентрация свободных радикалов (ROS) и менее выражены изменения в структуре митохондриальной ДНК. Таким образом, белок p66Shc служит важнейшим компонентом внутриклеточных окислительно-восстановительных процессов [34]. Хотя биохимическую роль белка p66Shc еще только предстоит точно установить, известно, что он участвует в митохондриальной продукции свободных радикалов, выступая в качестве окислительновосстановительного фермента, и способен окислять цитохром C, образуя проапоптотические свободные радикалы (ROS) в ответ на специфические стрессовые воздействия (рис. 14.6). Эти данные поддерживают концепцию центральной роли белка p66Shc в контролировании оксидативного стресса и участия его в патогенезе сосудистых заболеваний [35]. Экспрессия гена белка p66Shc значительно повышается в моноцитах крови больных сахарным диабетом, что
коррелирует с плазменной концентрацией изопростанов - маркеров оксидативного стресса in vivo [36]. Следовательно, генетическая делеция белка-адаптора p66 hc предотвращает индуцированную гипергликемией эндотелиальную дисфункцию и оксидативный стресс [37]. По совокупности указанных данных можно заключить, что белок p66Shc служит частью сигнального пути, относящегося к индуцированному гипергликемией сосудистому повреждению, и его можно рассматривать в качестве потенциальной мишени терапевтических воздействий, направленных против сосудистых осложнений сахарного диабета.
Рис. 14.6. Биохимические взаимодействия каскада белка p66Shc в митохондриальной электрон-транспортной цепи. Свободные радикалы активируют в-изоформу протеинкиназы C, индуцируя Se^-сериновое фосфорилирование белка p66Shc, которое позволяет перенести этот белок из цитозоля к митохондриям. В митохондриях p66Shc связывается с белками комплекса импорта TIM-TOM. Проапоптотические стимулы дестабилизируют комплекс p66Shc-mtHsp70 и приводят к переходу белка p66Shc в мономерную форму [8, 10]. Сразу после активации белок p66Shc начинает окислять цитохром С и катализирует превращение O2 в H2O2. Перекись водорода способствует открытию митохондриальных пор с последующим повышением проницаемости митохондриальной мембраны для ионов, растворимых веществ и воды, набуханием и разрушением органелл и дальнейшим высвобождением проапоптотических факторов в цитозоль. НАДН - никотинамидадениндинуклеотид (восстановленная форма), НАД+ - никотинамидадениндинуклеотид, ФАДН2 - флавинадениндинуклеотид Н2, ФАД - флавинадениндинуклеотид. Источник (с разрешения): Cosentino F., Francia P., Camici G.G. et al. Final common molecular pathways of aging and cardiovascular disease: role of the p66Shc protein // Arterioscler. Thromb. Vasc. Biol. - 2008. - Vol. 28. - P. 622-628.
Влияние сахарного диабета на сосудистую функцию не ограничивается воздействием на эндотелий. Происходит ослабление вазодилатирующего ответа на экзогенные доноры NO, а дисрегуляцию функции гладкомышечных клеток стенки сосуда еще более усугубляют нарушения функционирования симпатической нервной системы. При диабете увеличивается активность протеинкиназы C, продукция ядерного фактора-кВ и образование свободных кислородных радикалов, в том числе и в гладкомышечных клетках сосудов. Более того, диабет способствует усилению миграции гладкомышечных клеток сосудов в область формирующихся атеросклеротических повреждений, где эти клетки реплицируются и продуцируют внеклеточный матрикс. Так осуществляются важные этапы образования атеросклеротической бляшки [38]. Апоптоз сосудистых гладкомышечных клеток в атеросклеротической бляшке также усилен настолько, что в бляшках пациентов с диабетом обнаруживают малое количество гладкомышечных клеток, чем и обусловлена выраженная склонность бляшек к разрывам [39]. Выработка цитокинов приводит к уменьшению синтеза коллагена гладкомышечными клетками сосудов и повышает продукцию матричных металлопротеиназ, значительно повышая вероятность дестабилизации бляшки.
Учитывая вышеописанные эффекты гипергликемии на функцию сосудов, можно прийти к заключению, что обеспечение хорошего гликемического контроля само по себе способно защитить пациента от микро- и макрососудистого повреждения и улучшить прогноз. Эпидемиологические исследования доказали, что повышение плазменной концентрации глюкозы ассоциировано с сердечно-сосудистыми событиями. Однако об эффекте строгого гликемического контроля известно меньше. Несколько клинических исследований со значительными периодами наблюдения сосредоточились на изучении влияния различных сахароснижающих препаратов на смертность и развитие заболеваний системы крово-обращения у больных диабетом. Рандомизированное проспективное мультицентровое клиническое исследование UKPDS (The United Kingdom Prospective Diabetes Study, Проспективное исследование диабета в Великобритании) показало, что интенсивные схемы сахароснижающей терапии у пациентов с впервые выявленным сахарным диабетом типа 2 были ассоциированы с уменьшением риска микрососудистых осложнений и недостоверным снижением риска ИМ (p=0,052). Среди пациентов с избыточным весом, исходно получавших метформин, наблюдали снижение риска ИМ на 39% (p=0,01) и риска смерти от любой причины на 36% (p=0,01) [40]. При оценке состояния лиц, получавших лечение в рамках UKPDS, спустя 10 лет после завершения исследования сохранялось преимущество раннего достижения хорошего гликемического контроля в отношении микро- и макрососудистых исходов [41]. В группе больных, получавших препараты сульфонилмочевины и инсулин, относительное снижение риска сохранялось через 10 лет для любой связанной с диабетом конечной точки и микрососудистых поражений, а снижение риска
развития ИМ и смерти от любой причины появилось со временем, по мере возникновения сердечно-сосудистых событий. Долгосрочные преимущества после терапии метформином были выявлены в группе больных с избыточной массой тела (рис. 14.7). Риски и достоинства жесткого гликемического контроля будут подробнее обсуждены в следующих разделах.
Гипергликемия характерна как для диабета типа 1, так и для диабета типа 2. В нескольких исследованиях была выявлена связь между повышением концентрации глюкозы в плазме крови и возрастанием сердечно-сосудистой смертности и заболеваемости [14]. Растущие усилия направлены на выяснение влияния глюкозы на функцию сосудов, в том числе на эндотелиальную функцию и биодоступность оксида азота (NO).
Эндотелий вносит вклад в контроль тонуса гладкой мускулатуры стенки сосудов путем выделения NO, вызывающего вазодилатацию и ингибирующего тромбоциты (т.е. предотвращающего сосудистый спазм и формирование тромба).
Реакцию образования NO из терминального гуанидинового атома азота L-аргинина катализируют ферменты семейства NO-синтаз (NOSs). Один из этих ферментов, эндотелиальная NO-синтаза (eNOS), Ca2+-зависима. Она постоянно находится в клетках различных типов, включая клетки эндотелия.
Активность превращения L-аргинина в NO определяется балансом между синтезом и распадом NO путем его реакции с супероксид-анионом (O2-). В физиологических условиях O2- заметно не влияет на образование этой молекулы, поэтому NO может оказывать свое протективное действие на сосуды, способствуя поддержанию антиатеросклеротических условий. Однако при наличии факторов сердечно-сосудистого риска продукция супероксид-аниона быстро становится избыточной, NO инактивируется, повышается концентрация пероксинитрита (ONOO-) - очень мощного оксиданта (рис. 14.3).
итзидрргы j лт;р«иgt;еии |
образования супероксид-аниона [15-17] (рис. 14.4). Потеря биодоступности оксида азота предшествует развитию необратимого атеросклероза и служит независимым предиктором неблагоприятных сердечно-сосудистых событий [18].
Рис. 14.4. Гипергликемия и производимые эндотелием вазоактивные субстанции. ФНО - фактор некроза опухолей; НАДФ(Н+) - восстановленная форма
никотинамидадениндинуклеотидфосфата; цГМФ - циклический гуанозинмонофосфат; СОХ- 2 - циклооксигеназа-2; DAG - диацилглицерин; eNOS - эндотелиальная NO-синтаза; ЕТ - эндотелин;^-в - интерлейкины; МСР-1 - моноцитарный белок-хемоаттрактант-1; NFkB - нуклеарный фактор kB; PGIS - простациклинсинтаза; PGI2 - простациклин; РКС - протеинкиназа С; PLC - фосфолипаза С; ТхА2 - тромбоксан А2; VCAM-1 - сосудистые молекулы клеточной адгезии. Источник (с разрешения): Creager M.A., Luscher T.F.,
Cosentino F. et al. Diabetes and vascular disease: pathophysiology, clinical consequences and medical therapy: Part I // Circulation. - 2003. - Vol. 108. - P. 1527-1532.
Митохондриальная продукция O2- - важный медиатор гипергликемического поражения сосудов [16]. Дальнейшее повышение концентрации O2- происходит под действием порочного круга, в котором свободные кислородные радикалы (ROS - от reactive oxygen species, частицы активного кислорода) индуцируют активацию протеинкиназы C [15] и приводят к повышению протеинкиназа C-опосредованной продукции ROS. Показано, что активация протеинкиназы С глюкозой вовлечена в регуляцию и активацию НАД(Ф)^-зависимой оксидазы - важного сосудистого источника продукции O2- [16, 19]. Активность НАД(Ф)H+ и экспрессия субъединиц белка у пациентов с диабетом повышены во внутренних грудных артериях и больших подкожных венах бедра [20]. Хотя активация протеинкиназы С под действием высокой концентрации глюкозы ведет к ''up''-регуляции (т.е. к позитивной регуляции) экспрессии NO-синтазы, повышенный распад оксида азота перекрывает этот эффект и вызывает в конечном счете сокращение биодоступности NO. Таким образом, активация протеинкиназного пути - тот узел во внутриклеточной передаче сигнала, который ведет к индуцированному гипергликемией оксидативному стрессу и эндотелиальной дисфункции [21] (рис. 14.5).
Рис. 14.5. Объединенный протеинкиназа C-зависимый механизм служит триггером, при помощи которого гипергликемия индуцирует эндотелиальную дисфункцию и сосудистое воспаление. NO - оксид азота.
Избыток свободных кислородных радикалов влияет на эндотелиальную функцию несколькими путями.
Супероксид-анион быстро инактивирует NO и превращает его в пероксинитрит [22] - мощный оксидант, который легко проходит через фосфолипидные мембраны и вызывает нитрование субстратов, блокируя регуляторные рецепторы, ферменты-инактиваторы свободных радикалов [23, 24] и ключевые кофакторы эндотелиальной NO-синтазы (eNOS), например тетрагидробиоптерин [25].
Митохондриальная продукция супероксид-аниона повышает внутриклеточное образование продуктов конечного гликози-лирования, что неблагоприятно отра-жается на эндотелиальной функции в связи с усилением образования свободных радикалов и провоспалительных цитокинов в клетках сосудов и увеличением эндотелиальной экспрессии различных молекул адгезии, участвующих в атерогенезе [26].
Активация рецепторов продуктов конечного гликозилирования повышает внутриклеточное образование супероксид-аниона [27] и, вероятно, представляет собой ключевую ступень в развитии атеросклеротического поражения [28].
Продукция супероксид-аниона активирует гексозаминный путь, который снижает активацию NOS под действием протеинкиназы Akt [29]. Активация Akt далее ограничивается протеинкиназа С- зависимым ингибированием фосфатидилинозитол-3-киназного пути.
Оксидативный стресс в результате повышения концентрации глюкозы увеличивает содержание диметиларгинина - конкурентного антагониста NOS [30].
Белок-адаптор p66Shc контролирует клеточный ответ на оксидативный стресс. Для лабораторных крыс с дефицитом белка p66Shc(p66Shc-/-) характерна повышенная резистентность к свободным кислородным радикалам (ROS), большая продолжительность жизни и менее выраженный атеросклеротический процесс при высоком потреблении жиров. Это свидетельствует о положительном влиянии p66Shc на процесс старения и появление заболеваний с возрастом [31, 32]. Показано также, что у старых крыс с дефицитом белка p66Shc сохраняется, по сравнению с дикими сородичами того же возраста из того же помета, способность к эндотелийзависимой релаксации сосудов [33]. Кроме того, в клетках с фенотипом p66Shc-/- снижена внутриклеточная концентрация свободных радикалов (ROS) и менее выражены изменения в структуре митохондриальной ДНК. Таким образом, белок p66Shc служит важнейшим компонентом внутриклеточных окислительно-восстановительных процессов [34]. Хотя биохимическую роль белка p66Shc еще только предстоит точно установить, известно, что он участвует в митохондриальной продукции свободных радикалов, выступая в качестве окислительновосстановительного фермента, и способен окислять цитохром C, образуя проапоптотические свободные радикалы (ROS) в ответ на специфические стрессовые воздействия (рис. 14.6). Эти данные поддерживают концепцию центральной роли белка p66Shc в контролировании оксидативного стресса и участия его в патогенезе сосудистых заболеваний [35]. Экспрессия гена белка p66Shc значительно повышается в моноцитах крови больных сахарным диабетом, что
коррелирует с плазменной концентрацией изопростанов - маркеров оксидативного стресса in vivo [36]. Следовательно, генетическая делеция белка-адаптора p66 hc предотвращает индуцированную гипергликемией эндотелиальную дисфункцию и оксидативный стресс [37]. По совокупности указанных данных можно заключить, что белок p66Shc служит частью сигнального пути, относящегося к индуцированному гипергликемией сосудистому повреждению, и его можно рассматривать в качестве потенциальной мишени терапевтических воздействий, направленных против сосудистых осложнений сахарного диабета.
Рис. 14.6. Биохимические взаимодействия каскада белка p66Shc в митохондриальной электрон-транспортной цепи. Свободные радикалы активируют в-изоформу протеинкиназы C, индуцируя Se^-сериновое фосфорилирование белка p66Shc, которое позволяет перенести этот белок из цитозоля к митохондриям. В митохондриях p66Shc связывается с белками комплекса импорта TIM-TOM. Проапоптотические стимулы дестабилизируют комплекс p66Shc-mtHsp70 и приводят к переходу белка p66Shc в мономерную форму [8, 10]. Сразу после активации белок p66Shc начинает окислять цитохром С и катализирует превращение O2 в H2O2. Перекись водорода способствует открытию митохондриальных пор с последующим повышением проницаемости митохондриальной мембраны для ионов, растворимых веществ и воды, набуханием и разрушением органелл и дальнейшим высвобождением проапоптотических факторов в цитозоль. НАДН - никотинамидадениндинуклеотид (восстановленная форма), НАД+ - никотинамидадениндинуклеотид, ФАДН2 - флавинадениндинуклеотид Н2, ФАД - флавинадениндинуклеотид. Источник (с разрешения): Cosentino F., Francia P., Camici G.G. et al. Final common molecular pathways of aging and cardiovascular disease: role of the p66Shc protein // Arterioscler. Thromb. Vasc. Biol. - 2008. - Vol. 28. - P. 622-628.
Влияние сахарного диабета на сосудистую функцию не ограничивается воздействием на эндотелий. Происходит ослабление вазодилатирующего ответа на экзогенные доноры NO, а дисрегуляцию функции гладкомышечных клеток стенки сосуда еще более усугубляют нарушения функционирования симпатической нервной системы. При диабете увеличивается активность протеинкиназы C, продукция ядерного фактора-кВ и образование свободных кислородных радикалов, в том числе и в гладкомышечных клетках сосудов. Более того, диабет способствует усилению миграции гладкомышечных клеток сосудов в область формирующихся атеросклеротических повреждений, где эти клетки реплицируются и продуцируют внеклеточный матрикс. Так осуществляются важные этапы образования атеросклеротической бляшки [38]. Апоптоз сосудистых гладкомышечных клеток в атеросклеротической бляшке также усилен настолько, что в бляшках пациентов с диабетом обнаруживают малое количество гладкомышечных клеток, чем и обусловлена выраженная склонность бляшек к разрывам [39]. Выработка цитокинов приводит к уменьшению синтеза коллагена гладкомышечными клетками сосудов и повышает продукцию матричных металлопротеиназ, значительно повышая вероятность дестабилизации бляшки.
Учитывая вышеописанные эффекты гипергликемии на функцию сосудов, можно прийти к заключению, что обеспечение хорошего гликемического контроля само по себе способно защитить пациента от микро- и макрососудистого повреждения и улучшить прогноз. Эпидемиологические исследования доказали, что повышение плазменной концентрации глюкозы ассоциировано с сердечно-сосудистыми событиями. Однако об эффекте строгого гликемического контроля известно меньше. Несколько клинических исследований со значительными периодами наблюдения сосредоточились на изучении влияния различных сахароснижающих препаратов на смертность и развитие заболеваний системы крово-обращения у больных диабетом. Рандомизированное проспективное мультицентровое клиническое исследование UKPDS (The United Kingdom Prospective Diabetes Study, Проспективное исследование диабета в Великобритании) показало, что интенсивные схемы сахароснижающей терапии у пациентов с впервые выявленным сахарным диабетом типа 2 были ассоциированы с уменьшением риска микрососудистых осложнений и недостоверным снижением риска ИМ (p=0,052). Среди пациентов с избыточным весом, исходно получавших метформин, наблюдали снижение риска ИМ на 39% (p=0,01) и риска смерти от любой причины на 36% (p=0,01) [40]. При оценке состояния лиц, получавших лечение в рамках UKPDS, спустя 10 лет после завершения исследования сохранялось преимущество раннего достижения хорошего гликемического контроля в отношении микро- и макрососудистых исходов [41]. В группе больных, получавших препараты сульфонилмочевины и инсулин, относительное снижение риска сохранялось через 10 лет для любой связанной с диабетом конечной точки и микрососудистых поражений, а снижение риска
развития ИМ и смерти от любой причины появилось со временем, по мере возникновения сердечно-сосудистых событий. Долгосрочные преимущества после терапии метформином были выявлены в группе больных с избыточной массой тела (рис. 14.7). Риски и достоинства жесткого гликемического контроля будут подробнее обсуждены в следующих разделах.
Источник: Кэмм А. Джон, Люшер Томас Ф., Серруис П.В., «Болезни сердца и сосудов.Часть 3 (Главы 11-15)» 2011