
Апластическая анемия (АА) — заболевание, характеризующееся резким угнетением костномозгового кроветворения, торможением процессов пролиферации и дифференцировки клеточных элементов с развитием глубокой панцитопении в периферической крови. Апластическая анемия на протяжении многих десятков лет со времени первого описания, опубликованного в 1888 г. Р. Ehilich, для большинства больных была прогностически крайне неблагоприятным заболеванием. Частота встречаемости АА составляет
Апластические анемии могут быть наследственными и приобретенными. Наследственная форма АА (анемия Фанкони) сочетается с другими наследственными аномалиями. Приобретенные формы АА связаны с действием различных агентов: ионизирующая радиация, лекарственные препараты - антибиотики, сульфаниламидные, антитиреоидные, противосудорожные, противотуберкулезные, противодиабетические препараты, декарис, анальгин, « левомицетин, тетрациклин, бутадион, препараты золота и др., химические соединения (бензол и его производные, этилированный бензин, пестициды и др.), вирусы гепатита А, В, С, вирус Эпштейна-Барр, цитомегаловирус, вирусы герпеса, ВИЧ, парвовирус В19. Апластическая анемия, развивающаяся в течение первых 6 месяцев у больного, перенесшего острый вирусный гепатит, называют гепатитассоциированной АА. Доказана способность вирусов гепатита А, В, С ингибировать рост колоний и дифференцировку клеток- Предшсственников. Считается, что гепатитассоциированная АА, скорее всего, является следствием иммунной агрессии в отношении гемопоэза. Описаны ап-
ластические кризы у больных различными формами гемолитических анемий, вызванные парвовирусной инфекцией. Мишенью для парвовируса В19 служат эритроидные клетки-предшественники, поражение которых с врожденным Или приобретенным иммунодефицитом может привести к развитию парциальной красноклеточной аплазии. Из числа эндогенных факторов, угнетающих
гемопоэз, отмечают нарушение функции яичников, щитовидной и вилочковой желез. Изменение функции тимуса нередко сопровождается возникновением парциальной красноклеточной аплазии. В большинстве случаев АА этиологический фактор остается неизвестным {идиопатические АА).
Представления о патогенезе АА в 1970-80 гг. предполагали связь между развитием аплазии кроветворения и дефектом стволовых клеток, нарушением регуляции гемопоэза имму некомпетентны ми клетками и повреждением микроокружения, т. е. стромы костного мозга. Дефектность стволовых клеток при АА доказана культуральными исследованиями, в которых обнаружено снижение колониеобразующей способности клеток-предшественников гемопоэза. Иммунологическое фенотипирование клеток костного мозга выявило уменьшение количества клеток, экспрессирующих маркер CD34, характерный для ранних гемопоэтических клеток-предшественников. Функция стромы костного мозга у большинства больных АА страдает в меньшей степени, чем функция гемопоэтических клеток. Обнаружена нормальная или даже повышенная способность стромальных клеток при АА продуцировать гемопоэтические ростовые факторы. Только у небольшой части больных АА выявлена сниженная продукция стромальными клетками костного мозга ГМ-КСФ, ИЛ-3, ИЛ-1, ИЛ-6, при этом концентрация ЭПО и тромбопоэтина, Г-КСФ оказалась повышенной. Таким образом, многочисленными исследованиями с использованием культур клеток и молекулярно-биологического подхода не удалось доказать наличие при АА выраженного дефицита факторов роста и их ведущей роли в патогенезе АА.
В последние годы большинство исследователей ведущую роль в развитии АА придают иммунной деструкции гемопоэза. При АА показано повышение активности цитотоксических Т-лимфоцитов и натуральных киллеров, оказывающих подавляющее действие на костный мозг. Большинство фундаментальных исследований связывают развитие костномозговой недостаточности при АА с появлением в периферической крови и костном мозге активированных цитотоксических Т-лимфоцитов, продуцирующих фактор некроза опухоли (ФИО), у-интерферон, которые способны в условиях in vitro ингибировать нормальный гемопоэз. Вследствие этого происходит значительное снижение пула гемопоэтических клеток и развитие аплазии костного мозга.
В последние годы большое внимание уделяется изучению апоптоза как одного из механизмов развития аплазии кроветворения. При АА имеет место повышенная способность гемопоэтических клеток к апоптозу, т. к. на их поверхности экспрессируются рецепторы, являющиеся маркерами апоптоза (CD95). Воздействие на клетки ФНО, у-интерферона вызывает усиление экспрессии рецепторов апоптоза на клеточной мембране.
Исследования последних лет обнаружили наличие общего для пароксизмальной ночной гемоглобинурии и некоторых больных АА молекулярного дефекта, возникающего в результате соматической мутации гена PIG-A, связанного с Х-хромосомой и участвующего в синтезе белкового комплекса, называемого GPI (гликозилинозитолфосфолипидный якорь), способного инактивировать комплемент на поверхности клеток, в частности на поверхности эритроцитов. Высказывается предположение о том, что появление даже небольшого клона PIG-A дефектных гемопоэтических клеток может оказаться причиной иммунной атаки, направленной на нормальные кроветворные клетки и приводящей к развитию аплазии кроветворения.
Различные этиопатогенетические механизмы ведут к нарушению эритро-, грануло- и тромбоцитопоэза, что служит первоначальным антигенным сигналом, приводящим к активации иммунной системы или срыву толерантности и аутоиммунной деструкции гемопоэза при АА. В настоящее время АА рассматривается как гетерогенная по своему происхождению и механизмам развития группа аплазий кроветворения, для которых ведущими в патогенезе являются поражение стволовых клеток (первичное или в результате иммунной деструкции) и аутоиммунная агрессия в отношении гемопоэза (первичная или в ответ на появление клона дефектных стволовых клеток).
В патогенезе АА рассматривают следующие механизмы:
Клиническая картина определяется анемическим, геморрагическим синдромом и инфекционными осложнениями. Основные проявления АА обусловлены угнетением нормального кроветворения, гипоксией тканей и органов (одышка, тахикардия, слабость, головокружение) и резкой тромбо- цитопенией (кровоподтеки, петехии, носовые кровотечения, меноррагии и другие кровотечения). В результате выраженной нейтропении развиваются воспалительные процессы.
Критериями диагноза А А являются:
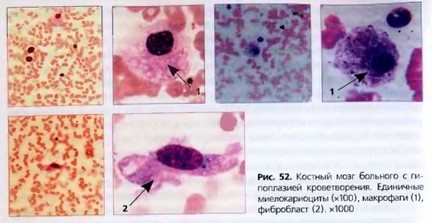
Костный мозг. Количество миелокариоцитов в костном мозге резко снижено (менее 40 х 109/л), отмечается замещение гемопоэтической ткани Жировой тканью. Наблюдается задержка созревания клеток трех ростков кроветворения. Обычно количество бластных клеток находится в пределах нормы. На фоне снижения общего числа гранулоцитов повышено относительное содержание лимфоцитов, плазматических клеток (до 10-12%). Встречаются макрофаги, липофаги (рис. 52). Эритропоэз характеризуется абсолютным уменьшением количества эритрокариоцитов и нарушением
их диффсренцировки. Количество сидеробластов и сидероцитов в костном мозге значительно возрастает. Резко снижено количество мегакариоцитов, а в тяжелых случаях они могут отсутствовать. При гистологическом исследовании трепанобионтатов костная ткань сохраняет нормальную структуру. Поражение костного мозга имеет очаговый характер. В местах опустошения активный костный мозг замещается жировой и/или фиброзной тканью. Однако даже при крайне тяжелом угнетении гемопоэза возможны активные очаги кроветворения.
Периферическая кровь. Характерны признаки выраженной нормохром- ной анемии с резким снижением концентрации НЬ (25-80 г/л), количества эритроцитов (0,7-2,5 х 1012/л), умеренным анизоцитозом с тенденцией к макроцитозу, пойкилоцитозом (рис. 53). Ретикулоцитопения варьирует от 0,3 до 0,9%, при гемолизе достигает 4-5%. Характерной для АА является выраженная лейкопения (до 2,5—0,55 х 109/л) с абсолютной нейтропенией и относительным лимфоцитозом. В случае присоединения инфекции может наблюдаться сдвиг влево до миелоцитов. Резко выражена тромбоцитопения (2,0-25,0 х 109/л), иногда в мазках периферической крови тромбоциты могут отсутствовать. В большинстве случаев АА ускорена СОЭ.
Тяжелая АА определяется при наличии двух любых из перечисленных критериев (гранулоцитопения lt;0,5 х 109/л, тромбоциты lt;20 х IOVjj, рети-

RBC 1,15 3.8-5,5 х 10,2/л
НЬ 37 130-155 г/л
Ht 12,2 36-48%
MCV 100,0 80-95 фл
МСН 32,2 27-31 пг
МСНС 322 300-380 г/л
RDW 25,9 11,5-14,5%
Рис. 53. Кровь больного с гипопластической анемией
кулоциты lt;] % с коррекцией по гематокриту или lt;20 х 109/л) в сочетании с аплазией КМ по данным трепанобиопсии. Сверхтяжелая АА - гранулоци- топения составляет lt;0,2 х 109/л.
На фоне частых гемотрансфузий при АА изменения в метаболизме железа характеризуются повышением содержания сывороточного железа и НТЖ что ведет к развитию гемосидероза. Увеличение концентрации эритропоэтина в крови связывают с падением утилизации его клеточными элементами костного мозга.
- на 1 млн в год, при колебании этого показателя в зависимости от страны от 0,6 до 5,0 на 1 млн в год.
Апластические анемии могут быть наследственными и приобретенными. Наследственная форма АА (анемия Фанкони) сочетается с другими наследственными аномалиями. Приобретенные формы АА связаны с действием различных агентов: ионизирующая радиация, лекарственные препараты - антибиотики, сульфаниламидные, антитиреоидные, противосудорожные, противотуберкулезные, противодиабетические препараты, декарис, анальгин, « левомицетин, тетрациклин, бутадион, препараты золота и др., химические соединения (бензол и его производные, этилированный бензин, пестициды и др.), вирусы гепатита А, В, С, вирус Эпштейна-Барр, цитомегаловирус, вирусы герпеса, ВИЧ, парвовирус В19. Апластическая анемия, развивающаяся в течение первых 6 месяцев у больного, перенесшего острый вирусный гепатит, называют гепатитассоциированной АА. Доказана способность вирусов гепатита А, В, С ингибировать рост колоний и дифференцировку клеток- Предшсственников. Считается, что гепатитассоциированная АА, скорее всего, является следствием иммунной агрессии в отношении гемопоэза. Описаны ап-
ластические кризы у больных различными формами гемолитических анемий, вызванные парвовирусной инфекцией. Мишенью для парвовируса В19 служат эритроидные клетки-предшественники, поражение которых с врожденным Или приобретенным иммунодефицитом может привести к развитию парциальной красноклеточной аплазии. Из числа эндогенных факторов, угнетающих
гемопоэз, отмечают нарушение функции яичников, щитовидной и вилочковой желез. Изменение функции тимуса нередко сопровождается возникновением парциальной красноклеточной аплазии. В большинстве случаев АА этиологический фактор остается неизвестным {идиопатические АА).
Представления о патогенезе АА в 1970-80 гг. предполагали связь между развитием аплазии кроветворения и дефектом стволовых клеток, нарушением регуляции гемопоэза имму некомпетентны ми клетками и повреждением микроокружения, т. е. стромы костного мозга. Дефектность стволовых клеток при АА доказана культуральными исследованиями, в которых обнаружено снижение колониеобразующей способности клеток-предшественников гемопоэза. Иммунологическое фенотипирование клеток костного мозга выявило уменьшение количества клеток, экспрессирующих маркер CD34, характерный для ранних гемопоэтических клеток-предшественников. Функция стромы костного мозга у большинства больных АА страдает в меньшей степени, чем функция гемопоэтических клеток. Обнаружена нормальная или даже повышенная способность стромальных клеток при АА продуцировать гемопоэтические ростовые факторы. Только у небольшой части больных АА выявлена сниженная продукция стромальными клетками костного мозга ГМ-КСФ, ИЛ-3, ИЛ-1, ИЛ-6, при этом концентрация ЭПО и тромбопоэтина, Г-КСФ оказалась повышенной. Таким образом, многочисленными исследованиями с использованием культур клеток и молекулярно-биологического подхода не удалось доказать наличие при АА выраженного дефицита факторов роста и их ведущей роли в патогенезе АА.
В последние годы большинство исследователей ведущую роль в развитии АА придают иммунной деструкции гемопоэза. При АА показано повышение активности цитотоксических Т-лимфоцитов и натуральных киллеров, оказывающих подавляющее действие на костный мозг. Большинство фундаментальных исследований связывают развитие костномозговой недостаточности при АА с появлением в периферической крови и костном мозге активированных цитотоксических Т-лимфоцитов, продуцирующих фактор некроза опухоли (ФИО), у-интерферон, которые способны в условиях in vitro ингибировать нормальный гемопоэз. Вследствие этого происходит значительное снижение пула гемопоэтических клеток и развитие аплазии костного мозга.
В последние годы большое внимание уделяется изучению апоптоза как одного из механизмов развития аплазии кроветворения. При АА имеет место повышенная способность гемопоэтических клеток к апоптозу, т. к. на их поверхности экспрессируются рецепторы, являющиеся маркерами апоптоза (CD95). Воздействие на клетки ФНО, у-интерферона вызывает усиление экспрессии рецепторов апоптоза на клеточной мембране.
Исследования последних лет обнаружили наличие общего для пароксизмальной ночной гемоглобинурии и некоторых больных АА молекулярного дефекта, возникающего в результате соматической мутации гена PIG-A, связанного с Х-хромосомой и участвующего в синтезе белкового комплекса, называемого GPI (гликозилинозитолфосфолипидный якорь), способного инактивировать комплемент на поверхности клеток, в частности на поверхности эритроцитов. Высказывается предположение о том, что появление даже небольшого клона PIG-A дефектных гемопоэтических клеток может оказаться причиной иммунной атаки, направленной на нормальные кроветворные клетки и приводящей к развитию аплазии кроветворения.
Различные этиопатогенетические механизмы ведут к нарушению эритро-, грануло- и тромбоцитопоэза, что служит первоначальным антигенным сигналом, приводящим к активации иммунной системы или срыву толерантности и аутоиммунной деструкции гемопоэза при АА. В настоящее время АА рассматривается как гетерогенная по своему происхождению и механизмам развития группа аплазий кроветворения, для которых ведущими в патогенезе являются поражение стволовых клеток (первичное или в результате иммунной деструкции) и аутоиммунная агрессия в отношении гемопоэза (первичная или в ответ на появление клона дефектных стволовых клеток).
В патогенезе АА рассматривают следующие механизмы:
- Дефект стволовых клеток вследствие воздействия неизвестного пускового агента (уменьшение числа клеток, экспрессирующих маркер CD34, характерный для ранних гемопоэтических клеток-предшественников, снижение колониеобразующей способности клеток-предшественников гемопоэза).
- Нарушение регуляции гемопоэза иммунокомпетентными клетками (повышение активности цитотоксических Т-лимфоцитов (CD8+), продуцирующих фактор некроза опухоли, у-интерферон, которые ингибируют нормальный гемопоэз и подавляют образование гемопоэтических колоний).
- Повреждение микроокружения, т. е. стромы костного мозга.
- Наследственный генетический дефект.
Клиническая картина определяется анемическим, геморрагическим синдромом и инфекционными осложнениями. Основные проявления АА обусловлены угнетением нормального кроветворения, гипоксией тканей и органов (одышка, тахикардия, слабость, головокружение) и резкой тромбо- цитопенией (кровоподтеки, петехии, носовые кровотечения, меноррагии и другие кровотечения). В результате выраженной нейтропении развиваются воспалительные процессы.
Критериями диагноза А А являются:
- трехростковая цитопения: анемия (НЬ lt;110 г/л), гранулоцитопения (гра- нулоциты lt;2,0 х Ю9/л), тромбоцитопения (тромбоциты lt;100 х 109/л);
- снижение клеточности костного мозга и отсутствие мегакариоцитов по данным пунктата костного мозга;
- преобладание жирового костного мозга по данным исследования тре- панобиоптата.
Костный мозг. Количество миелокариоцитов в костном мозге резко снижено (менее 40 х 109/л), отмечается замещение гемопоэтической ткани Жировой тканью. Наблюдается задержка созревания клеток трех ростков кроветворения. Обычно количество бластных клеток находится в пределах нормы. На фоне снижения общего числа гранулоцитов повышено относительное содержание лимфоцитов, плазматических клеток (до 10-12%). Встречаются макрофаги, липофаги (рис. 52). Эритропоэз характеризуется абсолютным уменьшением количества эритрокариоцитов и нарушением
их диффсренцировки. Количество сидеробластов и сидероцитов в костном мозге значительно возрастает. Резко снижено количество мегакариоцитов, а в тяжелых случаях они могут отсутствовать. При гистологическом исследовании трепанобионтатов костная ткань сохраняет нормальную структуру. Поражение костного мозга имеет очаговый характер. В местах опустошения активный костный мозг замещается жировой и/или фиброзной тканью. Однако даже при крайне тяжелом угнетении гемопоэза возможны активные очаги кроветворения.
Периферическая кровь. Характерны признаки выраженной нормохром- ной анемии с резким снижением концентрации НЬ (25-80 г/л), количества эритроцитов (0,7-2,5 х 1012/л), умеренным анизоцитозом с тенденцией к макроцитозу, пойкилоцитозом (рис. 53). Ретикулоцитопения варьирует от 0,3 до 0,9%, при гемолизе достигает 4-5%. Характерной для АА является выраженная лейкопения (до 2,5—0,55 х 109/л) с абсолютной нейтропенией и относительным лимфоцитозом. В случае присоединения инфекции может наблюдаться сдвиг влево до миелоцитов. Резко выражена тромбоцитопения (2,0-25,0 х 109/л), иногда в мазках периферической крови тромбоциты могут отсутствовать. В большинстве случаев АА ускорена СОЭ.
Тяжелая АА определяется при наличии двух любых из перечисленных критериев (гранулоцитопения lt;0,5 х 109/л, тромбоциты lt;20 х IOVjj, рети-
RBC 1,15 3.8-5,5 х 10,2/л
НЬ 37 130-155 г/л
Ht 12,2 36-48%
MCV 100,0 80-95 фл
МСН 32,2 27-31 пг
МСНС 322 300-380 г/л
RDW 25,9 11,5-14,5%
Рис. 53. Кровь больного с гипопластической анемией
кулоциты lt;] % с коррекцией по гематокриту или lt;20 х 109/л) в сочетании с аплазией КМ по данным трепанобиопсии. Сверхтяжелая АА - гранулоци- топения составляет lt;0,2 х 109/л.
На фоне частых гемотрансфузий при АА изменения в метаболизме железа характеризуются повышением содержания сывороточного железа и НТЖ что ведет к развитию гемосидероза. Увеличение концентрации эритропоэтина в крови связывают с падением утилизации его клеточными элементами костного мозга.