
Описано три механизма, регулирующих всасывание железа и, таким образом, поддерживающих баланс железа в организме:
- алиментарный регулятор,
- депо-регулятор,
- эритроидный регулятор.
Алиментарный регулятор: при высоком содержании в энтероцитах внутриклеточного железа подавляется экспрессия переносчика DMT-1 и клетки становятся резистентными к получению дополнительного железа. Этот регулятор реагирует только на алиментарное железо.
Депо-регулятор реагирует на общее содержание железа в организме, а не на алиментарное железо. В случае когда количество железа в депо (печень, селезенка, скелетные мышцы, эритроциты) падает, депо-регулятор увеличивает поступление железа в энтероциты до того момента, пока не восстановится его депо. Этот регулятор не оказывает значительного воздействия на всасывание гемового железа, а направлен на накопление негемового железа. Депо-регулятор также выполняет задачу предотвращения перегрузки железом после того, как потребности организма в железе обеспечены полностью. Он
перестраивает работу кишечного эпителия таким образом, что всасывание железа энтероцитами уменьшается в условиях повышенных запасов железа и, наоборот, повышает всасывание железа, когда потребности организма снижены. Предполагается, что ферритин, трансферрин и растворимый рецептор к трансферрину являются теми гуморальными факторами, которые передают сигналы от депо к кишечнику.
Эритроидный регулятор не реагирует на содержание железа в организме, а модулирует всасывание железа в ответ на потребности железа для эритропоэза. Эритроидный регулятор имеет более высокую способность для повышения абсорбции железа по сравнению с депо-регулятором. Так, при анемии абсорбция железа возрастает до 20-40 мг в день, что значительно выше тех величин, которые способен индуцировать депо-регулятор. Можно предположить, что эритрон должен оказывать влияние на всасывание железа в кишечнике, т. к. большая часть железа используется для нужд эритропоэза. Возможно, существует пока еще не выделенный гуморальный фактор, передающий сигналы из костного мозга в кишечник. Эритропоэтин таким фактором не является, т. к. в энтероцитах отсутствуют рецепторы к ЭПО.
Повышение всасывания алиментарного железа наблюдается при ЖДА, талассемиях, сидеробластных анемиях, наследственной дисэритропоэти- ческой анемии. В то же время при других формах анемий (наследственный сфероцитоз, АИГА, серповидноклеточная анемия), характеризующихся теми же изменениями эритропоэза, всасывание железа в кишечнике не стимулируется. Исходя из этого, гиперпролиферативные анемии разделяют на 2 класса: стимулирующие и не стимулирующие всасывание железа в кишечнике. При анемиях первого класса эритроидные клетки разрушаются внутри костного мозга (неэффективный эритропоэз), при анемиях второго класса эритроциты разрушаются в периферической крови. Значение места распада эритроцитов пока не определено.
Указанные регуляторы всасывания железа являются гуморальными факторами, они поддерживают гомеостаз железа в целом организме. Для поддержания гомеостаза железа в одной клетке существуют особые регуляторные механизмы. Единственной генетической системой регуляции являются изменения в экспрессии ферритина и рецепторов к трансферрину.
В последние годы уделяется большое внимание белку гепсидину (hepatic bactericidal protein), который является ключевым регулятором обмена железа. Эго гормон, состоящий из 25 аминокислотных пептидов, синтезируется преимущественно в печени, хотя низкая экспрессия его обнаружена в мышцах, кишечнике, желудке, легких и сердце, экскретируется с мочой. Синтез и секреция гепсидина контролируется тремя белками: HFE (белок наследственного гемохроматоза), гемоювелин, рецептор к трансферрину-2. Еепсидин обладает как антибактериальной активностью к грамотрицательным и грамположи- тельным бактериям, так и противогрибковой активностью.
Механизм действия гепсидина:
- ингибирует всасывание железа в кишечнике,
- блокирует транспорт железа через плаценту,
- блокирует выход железа из макрофагов.
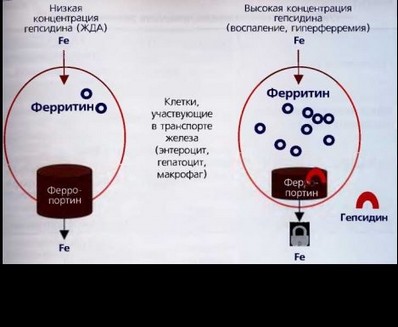
Молекулярной мишенью гепсидина является ферропортин (трансмембранный протеин), экспрессирующийся макрофагами, включая клетки Купфера, гепатоцитами, энтероцитами, клетками плаценты. Выход железа из клетки ингибируется связыванием гепсидина с ферропортином, последний погружается в цитоплазму и разрушается в лизосомах клетки. Таким образом, удаление ферропортина из базолатеральной мембраны приводит к блокаде выведения железа из клетки в плазму крови. Абсорбция железа энтероцитами осуществляется только в течение 2 дней, после чего они слущиваются и удаляются. Таким образом, транспорт железа ферропортином через базолатеральную мембрану приводит либо к связыванию с трансферрином плазмы, либо к выведению из организма со слущиваю- щимся эпителием кишечника. Когда запасы железа адекватны или высокие, синтезирующийся в печени гепсидин циркулирует в тонком кишечнике и вызывает интернализацию (погружение) ферропортина в цитоплазму клетки, блокируя единственный путь транспорта железа из энтероцита в плазму. При снижении запасов железа продукция гепсидина подавляется, ферропортин экспрессируется в большей концентрации на базолатеральной мембране энтероцита и других клеток, тем самым повышается выход железа из клетки в плазму и связывание его с трансферрином. Сходный механизм взаимодействия гепсидин - ферропортин объясняет присутствие макрофагов, перегруженных железом, которые часто встречаются при воспалительных заболеваниях, сопровождающихся высокой продукцией гепсидина. При высокой концентрации гепсидина блокируется ферропортин и, следовательно, выход железа из макрофагов.
Гепсидин быстро синтезируется в ответ на поступление алиментарного железа. В экспериментальных условиях инъекция одной дозы синтетического гепсидина мышам вызывала критическое падение концентрации железа в плазме крови в течение 1 часа. Таким образом, взаимодействие гепсидина с ферропортином создает нормальную концентрацию внеклеточного железа. Прямое взаимодействие гепсидина с ферропортином обеспечивает гомеостатический механизм, позволяющий регулировать уровень железа в плазме и его распределение в тканях. Механизм гиперпродукции гепсидина обусловлен воздействием провоспалительных цитокинов, преимущественно ИЛ-6, и наблюдается при воспалительных заболеваниях. Сниженная секреция гепсидина имеет место при ЖДА, гипоксии, неэффективном эритропоэзе (рис. 35).
Анемии, характеризующиеся перегрузкой железа, сопровождаются неэффективным эритропоэзом и повышением всасывания железа в тонком кишечнике. Наиболее часто такая анемия регистрируется при талассемиях. Парадоксальная ситуация возникает с синтезом гепсидина при талассемии. У этих больных концентрация гепсидина в моче низкая, несмотря на высокое содержание ферршина в сыворотке крови. Ингибицию гепсидина рассматривают как нецелесообразный физиологический ответ, который приводит к ухудшению перегрузки железом в тканях. Данный факт предположительно интерпретируется влиянием анемии на синтез гепсидина, ассоциированной с повышенным или неэффективным эритропоэзом. Низкий уровень гепсидина
при наследственных анемиях может быть одним из факторов, приводящих к гиперабсорбции железа, перегрузке и повреждению тканей, развитию фиброза.