
- Состояние БЦО при сепсисе можно охарактеризовать как обратимую недостаточность, в которую перерастает гипобиотическая или катаболическая неустойчивость, отличающие ССВО при воспалительных процессах. Численно, в соответствии со значением КИТС, бионедостаточносгь при сепсисе средней тяжести составляет в среднем 46%, что на 18% превосходит величину бионеустойчивости при ССВО. Бионедосгаточ- ность развивается при сепсисе в результате уменьшения КПп в среднем на 56% и КПд в среднем на 52% по сравнению с соответствующими базальными значениями этих параметров. Следует отметить, что по сравнению с ССВО величина КПп оказалась меньше в среднем на 22%, а КПд — на 25%. Указанные сдвиги с высокой степенью точности отражают снижение энергопроизводства и его ресурсообеспечения при сепсисе. Усугубляющийся энергодефицит прежде всего ответственен за уменьшение силовых характеристик сердца: в отсутствие пшоволе- мии САД понижался в среднем на 33%, а СИ — на 36% по сравнению с уровнем этих параметров, зарегистрированным при ССВО. Естественно, что указанные нарушения, в свою оче-
редь, ответственны за усугубление энергодефицита и бионедос- 5 таточности, так как вызывают тканевую ишемию, блокаду микроциркуляции (БМЦ) и эндотоксикоз.
- Возрастающая микробная нагрузка обусловлена перемещением бактерий из терминального отдела подвздошной кишки и слепой кишки, которые являются естественным резервуаром Гр(-) бактерий и других продуктов, содержащих эндотоксин. Целость слизистой оболочки кишки может быть нарушена при различных ситуациях, например таких, как блокада микроциркуляции, энтеральная недостаточность (ЭН) и воспалительные заболевания. Повреждение целости кишечника приводит к патологической проницаемости внутренней оболочки кишечника и может сопровождаться перемещением бактерий и/или эндотоксинов из ЖКТ в мезентериальные лимфатические сосуды и портальную систему. Дополнительными факторами этого процесса являются быстрое размножение кишечных бактерий и связанное с этим усиленное повреждение слизистой оболочки кишечника. В печени бактерии и эндотоксины могут запускать системный воспалительный ответ посредством активации Купферовских клеток и гепатоцитов. Если защитная система гепатоцитов не выполняет барьерной функции, бактерии и эндотоксины проникают в систему общей гемоциркуляции. Транслокация кишечной флоры может быть первичным или вторичным механизмом инициации и распространения системного воспалительного ответа, который обуславливает продолжение септического синдрома и развитие АС. У пациентов с септическим синдромом в 89% обнаружен эндотоксин в плазме.
При этом достоверно установлено, что большой процент эндотоксемии связан с процессом транслокации бактерий из просвета ЖКТ. ЭН и повышение внутрикишечного давления служат ускорению транслокации. В этом мы убедились совместно
- с С. И. Воротынцевым, когда на интактных крысах вызьшали летальный пневматоз внутрикишечным введением Ch. Через 2 ча- са от начала эксперимента в лимфоузлах брыжейки и в ткани печени обсемененность микрофлорой составляла 2106 КОЕ.
Развитие АС оказывается неизбежным, если транслокация микрофлоры составляет 10s и более.
Органоразобщающие механизмы ССВО являются основной причиной различных нарушений БЦО и развития АС. Общая схема органоразобщения, дисфункции и недостаточности БЦО представлены на рис. 5.1.
ссво
I
БМЦ
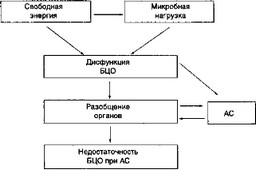
Рис. 5.1. Развитие бионедостаточности при АС
При нарастании микробной антигенной нагрузки нарушения БЦО определяются динамическим взаимодействием SIRS и CARS со следующими клиническими последствиями этого взаимодействия:
о превалирование SIRS-компонента выражается в кардио- 5 васкулярной компрометации (септический шок); органной дисфункции (моно- или мульти-); апоптозе (программированная клеточная гибель);
о превалирование CARS ведет к супрессии иммунной системы, что выражается в анергии и/или повышенной чувствительности к инфекции;
о сбалансированность SIRS и CARS — состояние восстановления БЦО (возврат к здоровью).
Необходимо отметить, что высокое персистирующее содержание антивоспалительных медиаторов предполагает неблагоприятный исход, так же как и длительное повышение концентрации провоспалительных медиаторов указывает на увеличение риска наступления летального исхода.
- Индукция оксида азота, согласно последним данным, в значительной мере модулирует течение SIRS, коррелируя с развитием септического шокового синдрома и несостоятельностью БЦО. Ему отводится центральная роль в падении давления крови, вызываемом эндотоксинами при септическом шоке. Введение эндотоксина разным видам лабораторных животных вызывает увеличение в крови и моче содержания нитрата — стабильного конечного продукта метаболических превращений N0.
У пациентов с септическим шоком регистрируются высокие концентрации нитрита-нитрата, которые кореллируют с падением сопротивления периферических сосудов. Последующие доказательства участия N0 в эндотоксиновом шоке были получены в экспериментах с использованием конкурентных ингибиторов ферментов, ответственных за образование N0: NO-синтаз (NOS), которые подавляли влияние эндотоксина на давление крови.
Описано существование двух фаз в гипотензивном ответе на эндотоксин:
- ° фаза немедленного падения давления крови, которая, оче
видно, обусловлена активацией конститутивной NOS (cNOS);
о фаза медленного снижения давления, начинающаяся через 90 мин после введения LPS. Эта фаза подавляется дексаметазо- ном или антителами против фактора некроза опухоли, что предполагает участие индуцибельной формы NOS (iNOS), на которую и влияет TNF.
Важная роль iNOS в действии эндотоксина следует также из прямых опытов по измерению активности iNOS в гомогенатах различных органов и тканей. Необходимо, однако, отметить, что помимо работ, в которых было показано повреждающее действие N0 на различные ткани, а также вызываемое N0 падение давления крови при действии эндотоксина, существуют также экспериментальные данные, свидетельствующие о том, что в некоторых условиях и при определенных концентрациях N0 может оказывать защитное действие.
Подавление образования N0 после введения эндотоксина сопровождается увеличением частоты клубочкового тромбоза, а также усилением повреждения кишечника и возрастанием сосудистой проницаемости.
- Эндотелий, подвергающийся воздействию цитокинов, усиливает:
о поверхностную адгезию молекул Е-селекгинов; о межклеточную адгезию молекул ICAM-1; о адгезию клеток сосудов VCAM (vascular cell adgesion molecule) для привлечения полиморфоядерных лимфоцитов и моноцитов с целью их прилипания и проникновения к очагу инфекции;
о изменение функций активированного эндотелия на проко- агулянтное и антифибринолитическое действие.
Решающее значение в скорости ферментативных процессов гемокоагуляции и объемах их протекания при АС принадлежит
количеству и активности тканевого фактора (тромбопластина). 5 Тромбин, возникающий в процессе свертывания крови, является наиболее мощным активатором тромбоцитов и патогенным инициатором локальных тромбозов. Он способен вызвать специфический протеолиз фибриногена с образованием фибрин- мономера. Последний, спонтанно полимеризуясь, образует трехмерную сеть — основу кровяного сгустка. Между свертыванием, которое приводит к откладыванию фибрина на сосудистой стенке, и фибринолизом, обеспечивающим растворение его, существует равновесие. Согласно теории физиологического внугрисосудистого микросвертывания крови, при физиологической травматизации кровеносных сосудов гемостатическая функция организма обеспечивает его нормальную жизнедеятельность благодаря ограничению и четкой локализации непрерывного внугрисосудистого свертывания крови. При массивной же тромбопластинемии саногенные реакции недостаточны для регуляции процессов полимеризации фибрин-мономера и олигомеров фибрина — растворимых комплексов фибрин-мономеров.
Не включившиеся в процесс полимеризации и унесенные током крови от места генерации тромбина, фибрин-мономер и дез-А- фибрин образуют олигомерные комплексы между собой, фибриногеном и фибронектином. Диссеменирование фибриновых эм- болов и растворимых комплексов фибрин-мономеров (РКФМ) происходит в результате распространения по циркуляции не только активных форм ферментов гемокоагуляционного каскада, но и продуктов самого клеточного распада. Возникающая полимеризация микроциркуляторного русла на фоне депрессии фиб- ринолитической активности крови инициирует отложение больших масс фибрина в сосудах, вызывая блок микроциркуляции, что ведет к ишемии тканей и органов. Альтерация клеток тканей сопровождается выбросом в кровоток новых порций тромбопластина, обуславливая новый каскад биохимических превращений.
- Морфологические исследования позволили выявить распро
страненный фибриновый тромбоз в капиллярах и венулах микро- циркуляторного русла легких, почек, геморрагические проявления, вторичные дистрофические и некротические изменения в «шоковых» органах. Патофизиологические нарушения, вызывающие и усугубляющие СОПЛ/РДСВ, представлены в табл. 5.1.
Таблица 5.1
Патофизиологические нарушения при СОПЛ/РДСВ
о Эндотелий задерживает агрегаты, нейтрофилы, тромбоциты, цитокины, др. медиаторы агрессии и повреждается.
о Часть агрессивных продуктов попадает в интерстиций легкого, а оттуда — в альвеолы.
о Массированная агрессия против эндотелия артериоспазм микротромбоз уменьшение продукции сурфактанта расширение повреждения альвеоло- капилярной мембраны дальнейшее ухудшение газообмена.
Распространенный фибриновый тромбоз микроциркулятор- ного русла при тромбогшастиновой коагулопатии отличался от тромбоза крупных сосудов. Для последних было характерно образование монолитных сгустков в просвете сосудов, состоящих из нитей фибрина (красный тип тромбоза) или тромбоцитов (белый тип тромбоза). Все это позволяет утверждать, что тром- бопластиновая коагулопатия — не нозологическая единица, а общепатологический неспецифический синдром, морфологические проявления которого являются одним из основных механизмов органоразобщения. Этот процесс лежит в основе синдромов невосстановленного кровотока, реперфузионного повреждения и кислородной задолженности. Продолжительность и распространенность его определяют выраженность функциональных и морфологических повреждений органов и тканей и, в конечном итоге, несостоятельность БЦО при АС.
- Маркером блокады микроциркуляции служит 5 ДВС. Первичным звеном патогенеза ДВС выступает попадание в сосудистое русло разнообразных активаторов свертывания крови, вызывающих агрегацию тромбоцитов и образование тромбина в циркулирующей крови. Это в свою очередь активирует, а значит и истощает свертывающую, калликреин-кинино- вую, комплемента и фибринолитическую ферментные системы крови. Циркуляция с кровью множества микросгустков и агрегатов форменных элементов обуславливает распространенную микроэмболизацию в микроциркуляторном русле. Это служит активирующим фактором образования распространенных микрогеморрагий, микротромбозов, возникновения во внутренних органах множественных микроскопических очагов некробиоти- ческих изменений и воспаления, лишенного биологической цели. В результате внутренние органы могут потерять критическое число своих анатомофункциональных единиц. Таким образом,
ДВС выступает фактором развития синдрома множественной системной недостаточности у больных в состоянии АС.
Начальным звеном патогенеза ДВС при септическом шоке служит повреждение эндотелия микрососудов при бактериемии и модуляции секреции эндотелиоцитами биоактивных веществ под влиянием возросшей действующей концентрации цитокинов в циркулирующей крови. Систему пара- и аутокринной регуляции микрососудов в структурном отношении составляют эндоте- лиоциты мельчайших артериол, секретирующие вещества, препятствующие агрегации тромбоцитов, простагландин 12, VIII фактор свертывания крови, активатор плазминогена с антикоагу- лянтной активностью, матричные белки и фибронекгин.
Повреждение бактериальными эндотоксинами эндотелия, стимуляция эндотелиоцитарной секреции в системе парааугок- ринной регуляции микрососудов приводит к активации фактора Хагемана, высвобождению тканевых тромбопластинов кле-
|
5. Эпсргоресуститация при сепсисе |
|
|
|
Таблица 5.2 |
|
Пути развития гиперкоагуляции |
|
- Активация внутреннего механизма свертывания крови.
При этом варианте развития гиперкоагулемии каскад коагуляции запускает активацию фактора Хагемана. Деструкция сосудистого эндотелия обуславливает прямой контакт циркулирующий крови с коллагеном соединительной ткани. Это ведет к трансформации предшественника XII фактора свертывания в активный фермент, индуцирующий алгоритм реакций свертывания в его ак- тивную форму.
- Активация внешнего механизма свертывания крови.
В результате бактериемической травматизации и вследствие адренергической стимуляции в ответ на снижение общего периферического сопротивления, эн- дотелиоциты микрососудов при АС начинают интенсивно высвобождать тромбопластин в циркулирующую кровь. Тромбопластин, взаимодействуя с кальцием и седьмым фактором свертывания, образует комплекс, активирующий фактор X. После этого этапа гиперкоагуляция продолжает развиваться в соответствии с униформным алгоритмом.
- Активация калликреиновой системы плазмы.
Активированный фактор Хагемана вызывает в плазме циркулирующей крови интенсивное образование фермента калликреина из прекалликреина. Кал- ликреин, в свою очередь, воздействует на свой главный субстрат — брадики- ниноген, который высвобождает нанопептид брадикинин, один из наиболее мощных естественных вазодилятаторов. У больных в состоянии АС, еще до активации калликреиновой системы под влиянием высокого содержания в крови цитокинов, общее периферическое сосудистое сопротивление снижено в результате артериоло-венулярного шунтирования. Расширение сосудов сопротивления при росте в плазме крови содержания брадикинина приводит к особенно быстрому и выраженному падению общего периферического сосудистого сопротивления и трудно купируемой артериальной гипотензии. Адренергическая стимуляция сосудистой стенки не в состоянии при септическом шоке восстановить общее периферическое сопротивление и артериальное давление, но усиливает интенсивное высвобождение тромбопластина эндоте- лиоцитами. Падение системного артериального давления ведет к циркуляторной гипоксии, повреждающей эндотелий на периферии, что способствует прямому контакту неактивированного фактора Хагемана с обнажившимся веществом соединительной ткани.
точными элементами микрососудов и агрегации тромбоцитов. Это, в свою очередь, активирует факторы свертывания крови.
Активация факторов свертывания происходит тремя путями 5 (табл. 5.2).
Все это превращает АС в самостоятельный фактор диссеминированного внутрисосудистого свертывания. Свертывание крови как защитная реакция, направленная на сохранение целости стенок сосудистого русла, во многом представляет собой результат взаимодействия между:
° эндотелиоцитоми;
о макромолекулами субэндотелиального слоя; о тромбоцитами и плазменными факторами свертывания. Патологические сдвиги в одном из трех эффекторов систем регуляции свертывания крови могут служить пусковым механизмом тромбогенеза. Ключевой фермент тромбогенеза и эффектор системной и локальной регуляции тромбообразования
- это тромбин. Тромбин является катализатором конечных этапов тромбоза (превращение фибрина в перекрёстно-связанный фибрин и активация фактора XIII свертывания крови). Активированный тромбином фактор XIII стабилизирует сгусток фибрина. Тромбин представляет собой высокоспецифичную сывороточную протеазу, обладающую биологической активностью, не связанной со свойствами тромбина как фермента. Так, относительно моноцитов и нейтрофилов тромбин выступает в качестве хемоаттрактанта и, соответственно, индуктора воспаления. Биологический смысл наличия у тромбина хемоаттрактивных свойств заключается в запуске воспаления через интенсивный тромбогенез в зоне первичной альтерации. Воспаление в данном контексте мы рассматриваем как защитно-патогенную системную реакцию, готовящую условия для последующего замещения дефектов тканей и органов через клеточную пролиферацию и ангиогенез. В этой связи становится ясной биологическая целесообразность свойств тромбина стимулировать клеточную пролиферацию и ангиогенез.
- Взаимодействие между антигенами и антителами может при
вести к образованию иммунных комплексов (ИК): растворимых и нерастворимых. Нерастворимые ИК способны откладываться в тканях и вызывать острые воспалительные реакции. Основные пути реализации этого воспаления представлены на рис. 5.2.
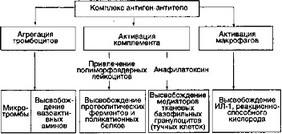
Рис. 5.2. Развитие воспалительной реакции под воздействием нерастворимых ИК
Тромбин, играя роль ключевого фермента тромбогенеза, одновременно представляет собой и индуктор активации молекулярных систем тромборезисгенгности эндотелия. Так, тромбин не только активирует факторы свертывания V и VIII, но и взаимодействует с белком наружной клеточной поверхности эвдогелиоцигов — тром- бомодулином, что ведет к резкой активации протеина С. Этот белок, перешедший в активированное состояние, инактивирует факторы свертывания V и VIII, что ограничивает образование фибрина Связывание тромбомодулином тромбина лишает тромбин способности превращать фибриноген в нерастворимый фибрин и блокирует реакцию активации тромбоцитов, а значит — и тромбообразование.
Протеин С — это естественный антикоагулянт, представляющий собой протеолотический фермент, синтез которого в печени проходит с участием витамина К. Свойствами антикоагулянта белок С обладает лишь в активированной форме. Так как
тромбомодулин находится лишь на поверхности интактных эн- 5 дотелиоцитов, то в поврежденном участке сосудистой стенки нет тромбомодулина и нет активной формы белка С. В результате в месте повреждения эндотелия и стенки сосуда свертывание крови и тромбогенез не испытывают ограничений. На поверхности прилежащих к локусу альтерации сосудистой стенки неповрежденных эндотелиальных клеток тромбомодулин есть, что ограничивает тромбообразование местом первичного повреждения стенки сосуда. Приобретенный дефицит белка С может быть причиной распространенного тромбоза и тромбоэмболии (табл. 5.3).
Таблица 5.3
Причины приобретенного дефицита белка С
- побочное действие антикоагулянтов кумаринового ряда
- интенсивная утилизация вследствие диссеминированного внутрисосудистого свертывания
- недостаточность печени
- респираторный дистресс-синдром взрослых
- послеродовая коагулопатия
- коагулопатия после интраоперационных кровотечений, тромбоз сосудов, что ведет к патогенно высокому потреблению белка С
- некоторые острые лейкемии
- миеломная болезнь
- системная красная волчанка и волчаночные иммуноглобулины-антикоагулянты, циркулирующие с кровью при расстройствах иммунной системы и инфекционных заболеваниях (синдромы первичного иммунодефицита и др.)
- побочное действие антибиотиков, молекула которых содержит N-метилтио- тетразоловое кольцо
- осложнение введения циклофосфамида, метатрексата
- потери антикоагулянтов с ультрафильтратом при гемодиализе и при нефротическом синдроме
Некоторые из ингибиторов протеаз вовлечены в регуляцию активности тромбина, циркулирующего с кровью. Наиболее мошный из известных естественных антикоагулянтов такого рода — это ангит- ромбин-UI, чьё сродство к тромбину усиливает гепарин. К факторам, противодействующим свертыванию крови, относят белки С и S. Агре- гащпо тромбоцитов тормозят просгациклин, фактор релаксации эндотелия, оксид азота. Некоторые из протеогликанов так же тормозят агрегацию тромбоцитов и их адгезию к эндотелию и субэндотелию.
- 5.1.6. Активные формы кислорода (АФК), ответствен
ные за развитие оксидантного стресса (ОС), обладают повреждающим действием. Основное количество кислорода, потребляемого клеткой, расходуется на энергетические нужды и используется в митохондриях. Связываясь с атомом железа цитох- ромоксидазы, молекула кислорода подвергается четырехэлектронному восстановлению и превращается в воду. Однако, наряду с митохондриальным окислением, в клетке протекает множество других окислительных процессов. Нарушение баланса между ними может негативно сказаться на метаболизме клетки и даже привести к ее гибели. Главной причиной негативных последствий от таких нарушений является образование, при неполном восстановлении кислорода, высокореакционных, а потому токсичных свободных радикалов или продуктов, их генерирующих. Семейство свободных радикалов характеризуется общим признаком — наличием на одном из атомов неспаренного электрона. Как правило, такое состояние веществ является неустойчивым и свободные радикалы стремятся превратиться в стабильные продукты путем спаривания свободного электрона. Это достигается либо путем отрыва атома (чаще всего атома
Н) от другого соединения и присоединения его к радикалу, либо за счет реакции рекомбинации, связанной с соединением двух радикалов в одну молекулу. Выбираемый способ самоуничтожения зависит от активности свободного радикала, то есть от его способности участвовать в отщеплении атома Н от соседних молекул. По этому признаку свободные радикалы условно делятся на активные и стабильные. Первые отличаются агрессивным поведением к своим соседям. Время их жизни невелико и они быстро исчезают, взаимодействуя с одной из близлежащих молекул. Как правило, результатом такого взаимодействия является отрыв атома Н и появление нового радикала. Такой процесс называют реакцией передачи цепи. Именно в результате такого
взаимодействия в клетке развиваются деструктивные процессы, 5 сопровождающиеся повреждением биологических мембран, молекул ДНК и белков, ответственных за нарушения биоустойчивости. Если активные свободные радикалы способствуют росту энтропии в биологических тканях, что ведет к развитию патологических состояний, ускоренному старению организма и его смерти, то стабильные радикалы тормозят развитие деструктивных процессов, замедляют старение и гибель клеток. Появление активных кислородосодержащих радикалов связано с процессами окислительного метаболизма. Поскольку интенсивность их протекания в биологических тканях неоднородна, то и зоны локального появления свободных радикалов очень гетерогенны. На уровне клетки до 60% всех выявленных радикалов образуются в митохондриях. Эго связано с тем, что данные органеллы являются главными энергопреобразующими системами клетки. Именно через них проходят основные потоки матаболизируемого кислорода. До 20% генерируемых в клетке радикалов образуются в микросомах, где осуществляется химическая переработка многих чужеродных для организма продуктов, включая лекарственные препараты. Оставшиеся 20% радикалов приходятся на остальные структуры клетки.
В настоящее время известна достаточно большая группа активных радикалов, появляющихся в биологических тканях.
К ним относятся супероксидный ион-радикал (О2*), гидроксильный радикал (НО*), пероксильный радикал (R02*). Специальными исследованиями удалось подтвердить факт появления алкильных радикалов (R ). Однако последние быстро связывают тканевой кислород и превращаются в более стабильные перок- сильные радикалы. Доказано образование алкоксильных (RO*) и хлороксильных (С10‘) радикалов.
Супероксидные ион-радикалы являются, как правило, первичными продуктами окислительно-восстановительных превращений. Современные методы исследования позволяют не
- только зафиксировать наличие в биологических объектах ион- радикалов Ог', но даже определить их концентрацию. В нормально метабилизирующих тканях концентрация таких молекул крайне низка и не превышает 1012 — 10 м М. Присутствие лишь следовых количеств СЬ‘ не должно смущать, поскольку такие частицы нестабильны и способны самопроизвольно диспропор- ционировать по реакции:
а+а+2н+ H2Q2 + O2.
В результате такой реакции образуется перекись водорода и регенерируется кислород, часть которого появляется в реакционной синглетной форме. Большая часть Ог* диспропорциониру- ет с участием специального фермента — супероксиддисмугазы (СОД).
Количество фиксируемой в тканях перекиси водорода достигает Ю'9—10'7 М, что на 3—4 порядка выше, чем Ог'. Это свидетельствует в первую очередь об относительной стабильности Н2О2 по сравнению с Ог*. Однако, в случае появления свободных ионов железа или меди, наблюдается быстрое разложение молекул Н2О2 по реакции Габера-Вейса с образованием высокореакционных радикалов НО*:
Н2О2 +Fe(2) НО+НО + Fe(3);
Fe(3) + 02' Fe(2) + О2.
Спектр возможных мишеней для гидроксильного радикала практически неограничен, но особенно любимым субстратом для гидроксильного радикала являются фосфолипиды, в состав которых входят полиненасьпценные жирные кислоты (ПНЖК). При этом, как правило, атом водорода отрывается от углерода, расположенного по соседству с двойной углерод-углеродной связью. Если молекулу фосфолипида условно представить в ви-
де LH, то реакция гидроксильного радикала с фосфолипидом 5 будет происходить по схеме:
LH + HO L+H2O.
Следует отметить, что если радикалы О2', НО* и синглетный кислород имеют малую молекулярную массу и могут участвовать в реакциях радикальной модификации по всей клетке, то алкильные и оксильные радикалы L‘ и L02* могут функционировать только в липидной фазе. Именно там протекают последовательно проходящие реакции, приводящие к накоплению гидроперекисей липидов в мембранах:
L' + Ch Ш' ; L(h‘ +LH LOOH + L' .
Количество гидроперекисей липидов (LOOH) может достигать 5 109 М/мг белка мембраны. Процесс ПОЛ чаще всего прекращается в результате взаимодействия пероксильного радикала с ингибитором свободно-радикальных реакций (LnH):
LO2' + LnH LOOH + Ln’ .
В результате этой реакции обрывается каскад реакций окисления ПНЖК, а образующийся малоактивный радикал Ln* быстро рекомбинирует.
В последние годы особое внимание уделяется продуктам взаимодействия малоактивного радикала азота (N'O), образующегося во многих тканях организма, с супероксидным ион-ради- калом. Продуктом реакции является пероксинитрит:
O2+NO + H* HOONO.
Последний представляет собой высокореакционноспобное соединение, инициирующее процессы ПОЛ в биомембранах и окисляющее белки и антиоксиданты.
- В другом ферментативном пути генерации супероксидного
радикала участвует ксантиноксидаза. Этот фермент катализирует реакции окисления гипоксантина в ксантин, а ксантина — в мочевую кислоту с образованием Ог‘: гипоксантин + 2 (к + НгО ксантин + 2Ch + 2Н*; ксантин + 2Ck + Н2О мочевая кислота + 20г’ + 2Н*.
В большинстве клеток основная масса фермента присутствует в виде NAD+ — зависимой ксантиндегидрогеназы, которая может превращаться в оксидазную форму специфичного окисления сульфгирильных групп белка или ограниченного протео- лиза Са2+-зависимой протеазой — каллеином.
Высокоспецифичная нейтрализация ‘О2* осуществляется ферментом супероксидциумутазой (СОД). Хотя дезактивация 'Ог* может достигаться путем спонтанной дисмутации, спонтанная реакция протекает слишком медленно, чтобы иметь физиологическое значение. Супероксиддисмутаза эффективно катализирует реакцию дисмутации 'Ог‘:
‘а+‘0г+2Н* H2O2 + Q2.
В условиях in vivo существуют и неферментативные пути образования АФК. В частности, именно такими путями возможно образование наиболее реакционно-способных из всех АФК. Т'/г для 'ОН в условиях живой клетки составляет всего лишь 109 секунды, поэтому первичное действие 'ОН проявляется в месте его генерации. Взаимодействие ‘ОН с окружающими молекулами приводит к образованию вторичных радикалов. Образование ‘ОН наблюдается в реакциях Габера-Вейса при взаимодействии 'Ог' с Н2О2 в присутствии ионов металла переменной валентности (обычно железа или меди):
Q2+H2Q2 О2 + ОН + 0Н\
Есть основания полагать, что в воспаленных и поврежден- 5 ных тканях создаются более благоприятные условия для протекания такой реакции, чем в интактных тканях.
Образование 'ОН в биологических средах возможно также в реакции Фентона — взаимодействие Н2О2 с Fe2+:
Fe2+ + Н2О2 Fe3+ + ОН + ОН".
Кроме того, полагают, что ОН может генерироваться нейтро- филами и моноцитами человека по механизмам, зависимым от миелопероксидазы.
Генерация N0 часто происходит совместно с образованием ’О2’. Эти радикалы с высокой скоростью взаимодействуют между собой (константа скорости 6.7х 109М‘|С"1) с образованием пе- роксинитрита (0N00‘). Вероятно, именно с пероксинитритом связано повреждающее действие N0 на биологические макромолекулы, в первую очередь на белки.
Пероксильные радикалы представляют собой промежуточные формы, образующиеся в процессе цепных реакций пере- кисного окисления липидов. Пероксид водорода не является свободно-радикальным соединением. Молекула Н2О2 способна проникать через мембраны и взаимодействовать с различными компонентами, локализованными внутри субклеточных структур. В условиях in vivo Н2О2 образуется в основном в результате дисмутации 'О2' и в реакциях, катализируемых оксидазами. При низких (микромолярных) концентрациях окислительная способность Н2О2 не велика.
Защита от повреждающего действия H2OV обеспечивается ферментом каталазой. Этот фермент с высокой скоростью превращает Н2О2 в О2 и Н2О:
Н2О2 + Н2О2 2Н2О2 + Ог.
- Сильным окислительным действием обладает гипохлорная
кислота (НСЮ). Этот вид АФК, как и Н2О2, не является свободным радикалом. НСЮ в значительных количествах продуцируется ферментом активированных нейтрофилов — миелоперок- сидазой, катализирующей окисление СГ в присутствии Н2О2. Установлено, что при содержании в 1 мл 5х 106 активированных нейтрофилов человека концентрация НСЮ за 2 часа достигает 88 мкМ, причем на образование НСЮ используется до 30% поглощаемого клетками кислорода. Кроме окислительной активности, НСЮ обладает сильным хлорирующим действием. В частности, входящий в состав клеточных мембран холестерин превращается под действием НСЮ в холестеинхлоргидрин, что ведет к нарушению целости мембран и лизису клеток.
В качестве основных компонентов неферментной системы защиты клеток человека от АФК можно назвать восстановленный глугатион, витамин С (аскорбиновая кислота), витамин Е (а-токоферол) и другие токоферолы. Кроме того, определенное защитное действие проявляют также мочевая кислота, аминокислота таурин и особенно белки, содержащие ионы металлов переменной валентности — трансферрин, лактоферрин, церулоплазмин и альбумин. Связывая ионы металлов, такие белки снижают вероятность протекания ‘ОН-генерирующих реакций, например реакции Фентона. Возможно, что функцию защиты от АФК, особенно ‘ОН, выполняют также высокомолекулярные гликопротеины слизистой трахеобронхиального дерева и желудочно-кишечного тракта.
Одним из наиболее изученных компонентов клетки, весьма чувствительным к модифицирующему действию свободно-ра- дикальных форм АФК, являются входящие в состав липидов ненасыщенные жирные кислоты. Процессы ПОЛ постоянно протекают во всех клетках в норме, являясь, вероятно, одним из способов их метаболической регуляции. При различных патоло-
гических состояниях, сопровождающихся некомпенсированной 5 генерацией свободных радикалов, изменяется характер ПОЛ, что приводит к неблагоприятным последствиям для интактной клетки. С одной стороны, интенсивные процессы ПОЛ приводят к модификации мембранных липидов, следствием чего является уменьшение текучести мембраны и увеличение проницаемости мембраны для различных ионов. В отдельных случаях может происходить нарушение целости мембраны, приводящее к таким нежелательным последствиям, как выход внутриклеточных компонентов, например гидролитических ферментов лизо- сом, в окружающую среду. С другой стороны, образующиеся при ПОЛ гидроперекиси и продукты их распада цитотоксичны.
Первичная модификация клеточных мембран под действием АФК дает различные вторичные эффекты. Одним из таких эффектов, оказывающим большое влияние на функционирование клетки, является нарушение гомеостаза Са2+. В нормальных условиях содержание Са2+ внутри клетки намного (до 104 раз) ниже, чем во внеклеточном пространстве. Кроме того, субклеточные структуры имеют существенно различное содержание Са2+.
Изменения проницаемости клеточных мембран а результате модифицирующего действия АФК приводит к изменению внутриклеточной концентрации Са2+, в частности резко увеличивается его содержание в цитоплазме. Это вызывает активацию Са2+-зависимых протеаз и фосфолипазы Аг. Неконтролируемый протеолиз вызывает острое повреждение клетки. Кроме того, протеолитическая модификация приводит к активации других протеолитических ферментов, например каллеина, трансформирующего ксантиндегидрогеназу в ксантиноксидазу. В результате такой трансформации стимулируется вторичное образование АФК.
Активация Са2+ фосфолипазы Аг способствует освобождению арахидоновой кислоты из мембранных фосфолипидов. Это
- вызывает запуск «каскада арахидоновой кислоты», в котором происходит синтез простагландинов, тромбоксанов и лейкотри- енов. Последние способны вызывать адгезию лейкоцитов к эндотелию и их активацию. Активация лейкоцитов, как отмечалось выше, приводит к образованию ‘О2'.
Продукты окислительной модификации белков на всех структурных уровнях зависят от природы АФК. Например, остатки тирозина иод действием гидроксильных радикалов трансформируются в о-дитирозин и L-диоксифенилаланин; N0 и пероксинитрит превращает их в 3-нитротирозин. Дня большинства белков интенсивное воздействие радикалов ОН приводит к их агрегации, но в присутствии ‘02‘ предпочтительным процессом становится фрагментация белковых макромолекул. Среди белков, наиболее чувствительных к модифицирующему воздействию гидроксильных радикалов, следует в первую очередь указать сывороточный альбумин. Константа скорости реакции ОН с этим белком более 1010M lc l.
Интенсивное модифицирующее действие на белки оказывает гипохлорит. Вероятно, первичными центрами такой модификации являются серу-содержащие (цистеин, метионин и цистин) и ароматические (триптофан, тирозин и гистидин) аминокислотные остатки, что затем приводит к структурным изменениям и фрагментации белков. Из основных белков плазмы крови особенно чувствительным к окислению в присутствии гипохлорита оказался церулоплазмин.
Нитрозилирование сулъфгидрильных центров в белках является наиболее характерной особенностью модифицирующего действия оксида азота и пероксинитрита. Кроме того, эти АФК эффективно взаимодействуют с остатками тирозина и лизина белков. По сравнению с другими АФК, перокс