ГЕНЕТИКА И ПАТОФИЗИОЛОГИЯ
Число генов, способных вызывать удлинение интервала Q-T и нарушения ритма, постоянно растет. Интересно, что различные варианты синдрома включают фенотипы, отличные от удлиненного интервала Q-T, аритмий и ВСС. Кроме того, хотя синдром удлиненного интервала Q- T изначально считался изолированной сердечной каналопатией, на сегодняшний момент ясно, что к развитию этого заболевания могут приводить и гены, не кодирующие ионные каналы. С другой стороны, все еще актуальной является концепция, полагающая, что гены, ответственные за синдром удлиненного интервала Q-T, влияют на ионные токи непосредственно (мутации ионных каналов) или опосредованно (белки-шапероны и другие модуляторы). Мутации в генах, кодирующих калиевые каналы, таких как KCNQ1 (синдром удлиненного интервала Q-T, вариант 1) и KCNH2 (вариант 2), и в гене, кодирующем натриевый канал SCN5A (вариант 3), были обнаружены первыми среди генетических причин синдрома удлиненного интервала Q-T. Варианты синдрома удлиненного интервала 1-3 составляют более 90% всех случаев данной патологии с известным генотипом [38], поэтому большая когорта пациентов, страдающих одним из трех вариантов этого заболевания, стала доступна для исследований корреляций генотипа и фенотипа.
В настоящее время определены 12 генов синдрома удлиненного интервала Q-T (см. табл. 9.6, рис. 9.4). Некоторые из этих генов (кодирующие белки анкирин-В, кавеолин-3, белки, фиксирующие А- киназу и синтропин) вызывают синдром удлиненного интервала Q-T, изменяя внутриклеточную локализацию белка, пропускную способность ионных каналов, ответ на симпатическую стимуляцию или нитрозилирование ионного канала [41-43]. Другие гены вызывают внесердечные нарушения. Так, мутация CACNA1C вызывает синдром Тимоти - высоколетальный и редкий вариант, включающий сочетание удлиненного интервала Q-T с желудочковыми аритмиями, врожденными пороками сердца (открытое овальное окно, тетрада Фалло), нарушениями развития, аутизмом и нарушениями развития лицевого скелета. Мутация KCNJ2 вызывает синдром
Андерсена: удлиненный интервала Q-T, аритмии, преходящий паралич и нарушения формирования лицевого скелета (см. табл. 9.6).
ОЦЕНКА РИСКА И ЛЕЧЕНИЕ
В последние несколько лет были очерчены различные свойства трех наиболее часто встречающихся аутосомно-доминантных варианта синдрома удлиненного интервала Q-T (LQTS 13). Корреляция генотип-фенотип для остальных генов часто может быть сведена к результирующему дефекту ионного тока (табл. 9.7).
Таблица 9.7. Генетические локусы и гены, участвующие в развитии аритмогенных заболеваний
продемонстрированы локус-специфичные различия, что позволило проводить оценку риска, основанную на генотипе [44]. Наихудший прогноз связан с длительностью интервала Q-T более 500 мс и генотипом вариантов 2 и 3. Пол пациента по-разному влияет на исход заболевания в зависимости от генетического дефекта: подгруппы наиболее высокого риска составляют мужчины с вариантом 3 и женщины с вариантом 2.
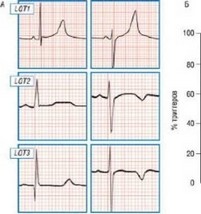 Е ФчЛнвькая могруэм
Е ФчЛнвькая могруэм
¦ ЭнщионщьниЯ арвсс В Lu-i. С^стямне мкн^ВД
tart LOTS LOT}
Рис. 9.5. Корреляция генотипа-фенотипа при синдроме удлиненного интервала Q-T: морфология реполяризации, вероятность события, триггеры и ответ на терапию в- адреноблокаторами в трех наиболее распространенных генетических вариантах (1-3) синдрома удлиненного интервала Q-T.
В 2004 г. были доложены данные по чувствительности к терапии пациентов с синдромом удлиненного интервала Q-T, входящих в большие когорты, наблюдаемые фондом Могери (Maugeri) в Италии. У пациентов с вариантом 1 синдрома отмечался очень хороший ответ на терапию p-адреноблокаторами, поскольку 90% из них не переносили обморок или остановку сердца в течение последующих 5,4 года наблюдения и продемонстрировали общую смертность, равную 1% [45]. Небольшому количеству больных с вариантом 1, получающих р-адрено- блокаторы, потребовалось дополнительное лечение: например, ИКД или левосторонняя десимпатизация сердца (левая каротидная симпатическая денервация). Обращает на себя внимание тот факт, что наиболее важной причиной кардиальных событий, возникающих во время антиадренергического лечения больных с вариантом 1 синдрома удлиненного интервала Q-T, является недостаточная комплаентность [46].
Посредством мультивариантного анализа также были определены предикторы кардиальных событий во время антиадренергической терапии:
удлинение интервала Q-T gt;500 мс;
возникновение жизнеугрожающих аритмий в возрасте до 7 лет;
наличие у пациента вариантов 2 и 3 синдрома.
Носители мутаций KCNH2 или SCN5A имеют относительный риск развития жизнеугрожающих аритмий на фоне приема p-адреноблокаторов, равный 2,81 и 4 соответственно, по сравнению с больными с вариантом 1 синдрома удлиненного интервала Q-T [45]. В соответствии с руководством Американского кардиологического колледжа, Американской ассоциации кардиологов и Европейского общества кардиологов (ACC/AHA/ESC) от 2006 г. [1], использование ИКД у больных с вариантами 2 и 3 (интервал Q-T gt;500 мс) в качестве основного средства профилактики остановки сердца имеет класс доказательности IIB. Поскольку ИКД не предотвращает развитие
аритмий и учитывая молодой возраст пациентов, его имплантация предрасполагает к осложнениям, связанным с разрывом отводящих электродов и ложным срабатыванием. Соответственно поиск терапевтических альтернатив является приоритетным в данной области исследований.
В настоящее время определены 12 генов синдрома удлиненного интервала Q-T (см. табл. 9.6, рис. 9.4). Некоторые из этих генов (кодирующие белки анкирин-В, кавеолин-3, белки, фиксирующие А- киназу и синтропин) вызывают синдром удлиненного интервала Q-T, изменяя внутриклеточную локализацию белка, пропускную способность ионных каналов, ответ на симпатическую стимуляцию или нитрозилирование ионного канала [41-43]. Другие гены вызывают внесердечные нарушения. Так, мутация CACNA1C вызывает синдром Тимоти - высоколетальный и редкий вариант, включающий сочетание удлиненного интервала Q-T с желудочковыми аритмиями, врожденными пороками сердца (открытое овальное окно, тетрада Фалло), нарушениями развития, аутизмом и нарушениями развития лицевого скелета. Мутация KCNJ2 вызывает синдром
Андерсена: удлиненный интервала Q-T, аритмии, преходящий паралич и нарушения формирования лицевого скелета (см. табл. 9.6).
ОЦЕНКА РИСКА И ЛЕЧЕНИЕ
В последние несколько лет были очерчены различные свойства трех наиболее часто встречающихся аутосомно-доминантных варианта синдрома удлиненного интервала Q-T (LQTS 13). Корреляция генотип-фенотип для остальных генов часто может быть сведена к результирующему дефекту ионного тока (табл. 9.7).
Таблица 9.7. Генетические локусы и гены, участвующие в развитии аритмогенных заболеваний
|
Названи е гена |
Название локуса |
Локус хромосо мы |
Тип наследован ия |
Белок |
Функциональны й эффект |
Фенотип |
|
Названи е гена |
Название локуса |
Локус хромосо мы |
Тип наследован ия |
Белок |
Функциональны й эффект |
Фенотип |
|
KCNQ1 |
LQTS1 |
11 p15.5 |
Аутосомно- доминантн ый |
IKs а- субъединица калиевого канала |
Потеря функций |
Удлиненный интервал Q-T |
|
|
JLN1 |
|
Аутосомно- рецессивн ый |
(KvLQT 1) |
Потеря функций |
Удлиненный интервал Q-T, глухота |
|
|
SQT2 |
|
Аутосомно- доминантн ый |
|
Приобретение функций |
Укороченный интервал Q-T |
|
|
AF1 |
|
Аутосомно- доминантн ый |
|
Приобретение функций |
ФП |
|
KCNH2 |
LQTS2 |
7q35-q36 |
Аутосомно- доминантн ый |
W а- субъединица калиевого канала |
Потеря функций |
Удлиненный интервал Q-T |
|
|
SQT1 |
|
Аутосомно- доминантн ый |
(HERG) |
Приобретение функций |
Укороченный интервал Q-T |
|
|
AF2 |
|
Аутосомно- доминантн ый |
|
Приобретение функций |
ФП |
|
SCN5A |
LQT3 |
3p21 |
Аутосомно- доминантн ый |
Сердечная а- субъединица натриевого канала (Nav 1.5) |
Приобретение функций |
Удлиненный интервал Q-T |
|
|
BrS1 |
|
Аутосомно- доминантн ый |
Потеря функций |
Синдром Бругада |
|
|
|
AF3 |
|
Аутосомно- доминантн |
|
Потеря |
ФП |
|
|
|
|
ый |
|
функций |
|
|
|
PCCD |
|
Аутосомно- доминантн ый |
|
Потеря функций |
Дефект системы проведения |
|
|
SSS |
|
Аутосомно- доминантн ый |
|
Потеря функций |
СССУ |
|
KCNJ2[2] |
AND/LQT S7 |
17q23.1- q24.2 |
Аутосомно- доминантн ый |
IK1 калиевый канал (Kir2.1) |
Потеря функций |
Удлиненный интервал Q-T, калий- чувствительный периодический паралич, дисморфия |
|
|
SQT3 |
|
Аутосомно- доминантн ый |
|
Приобретение функций |
Укороченный интервал Q-T |
|
|
AF4 |
|
Аутосомно- доминантн ый |
|
Приобретение функций |
ФП |
|
KCNE1 |
LQTS5 |
21q22.1- q22.2 |
Аутосомно- доминантн ый |
IKs Р- субъединица калиевого канала (MinK) |
Потеря функций |
Удлиненный интервал Q-T |
|
|
JLN2 |
|
Аутосомно- рецессивн ый |
Потеря функций |
Удлиненный интервал Q-T, глухота |
|
|
|
AF5 |
|
Аутосомно- доминантн ый |
|
Приобретение функций |
ФП |
|
ANK2* |
LQTS4 |
4q25-q27 |
Аутосомно- доминантн ый |
Анкирин B, заякоривающ ий белок |
Потеря функций |
Удлиненный интервал Q-T, ФП |
|
KCNE2 |
LQTS6 |
21q22.1- q22.2 |
Аутосомно- доминантн ый |
Ikt Р- субъединица калиевого канала |
Потеря функций |
Удлиненный интервал Q-T |
|
|
AF6 |
|
Аутосомно- доминантн ый |
(MiRP) |
Приобретение функций |
- |
|
CACNA 1C |
TS/LQTS 8 |
12p13.3 |
Аутосомно- доминантн ый /мозаицизм |
а- Субъединица кальциевого канала |
Приобретение функций |
Синдром Тимоти: удлиненный интервал Q-T, синдактилия, дефекты перегородки, ДМПП |
|
|
BrS3 |
|
Аутосомно- доминантн ый |
|
Потеря функций |
Синдром Бругада с укороченным интервалом Q-T |
|
CACNB 2B |
BrS4 |
10p12 |
Аутосомно- доминантн ый |
в- Субъединица кальциевого канала |
Приобретение функций |
Синдром Бругада с укороченным интервалом Q-T |
|
Cav3 |
LQTS9 |
3p24 |
Аутосомно- доминантн ый |
Кавеолин |
Приобретение функций тока натрия |
Удлиненный интервал Q-T |
|
SCNb4 |
LQTS10 |
11 q23.3 |
Аутосомно- доминантн ый |
в- Субъединица натриевого канала |
Приобретение функций тока натрия |
Удлиненный интервал Q-T |
|
AKAP9 (yotiao) |
LQTS11 |
7q21-q22 |
Аутосомно- доминантн ый |
A-киназа заякоривающ его белка |
Сниженный ток калия из-за потери чувствительнос ти к кАМФ |
Удлиненный интервал Q-T |
|
SNTA1 |
LQTS12 |
20q11.2 |
Аутосомно- доминантн ый |
а1- Синтрофин |
Увеличенный ток натрия благодаря S- нитрозилирова нию натриевых каналов SCN5A |
Удлиненный интервал Q-T |
|
GPD1-L |
BrS2 |
3p22.3 |
Аутосомно- доминантн ый |
Глицерол-3- фосфат дегидрогеназ а-1 - подобный белок |
Сниженный ток натрия |
Синдром Бругада |
|
RyR2 |
CPVT1 |
1q42-43 |
Аутосомно- доминантн ый |
Сердечный рианадиновы й рецептор |
Выброс кальция в диастолу |
Катехоламинергиче ская тахикардия |
|
CASQ2 |
CPVT2 |
1p13.3- p11 |
Аутосомно- рецессивн ый |
Сердечный кальсеквестр ин |
Выброс кальция в диастолу |
Катехоламинергиче ская тахикардия |
продемонстрированы локус-специфичные различия, что позволило проводить оценку риска, основанную на генотипе [44]. Наихудший прогноз связан с длительностью интервала Q-T более 500 мс и генотипом вариантов 2 и 3. Пол пациента по-разному влияет на исход заболевания в зависимости от генетического дефекта: подгруппы наиболее высокого риска составляют мужчины с вариантом 3 и женщины с вариантом 2.
Е ФчЛнвькая могруэм
¦ ЭнщионщьниЯ арвсс В Lu-i. С^стямне мкн^ВД
tart LOTS LOT}
|
|
атс |
Ппквтрантнол’ь ft) |
осгэновка Ёордда/шндЗагтия ej [MB * mi Смерть (lt;Ь) |
случаи до тарами Р-адомо Опакзгпрлчи fit) |
СЛуНЗИ Т4СПС чрвпикр-ад-роно- ОМЖЭТДОШ (VI |
|
UH7 |
45Г t нм: |
as |
to |
39 |
г |
|
LQT2 |
4Liг t им; |
ГО |
20 |
Л |
в |
|
ЮТЗ |
m f юие |
re |
|
5Г |
XI |
Рис. 9.5. Корреляция генотипа-фенотипа при синдроме удлиненного интервала Q-T: морфология реполяризации, вероятность события, триггеры и ответ на терапию в- адреноблокаторами в трех наиболее распространенных генетических вариантах (1-3) синдрома удлиненного интервала Q-T.
В 2004 г. были доложены данные по чувствительности к терапии пациентов с синдромом удлиненного интервала Q-T, входящих в большие когорты, наблюдаемые фондом Могери (Maugeri) в Италии. У пациентов с вариантом 1 синдрома отмечался очень хороший ответ на терапию p-адреноблокаторами, поскольку 90% из них не переносили обморок или остановку сердца в течение последующих 5,4 года наблюдения и продемонстрировали общую смертность, равную 1% [45]. Небольшому количеству больных с вариантом 1, получающих р-адрено- блокаторы, потребовалось дополнительное лечение: например, ИКД или левосторонняя десимпатизация сердца (левая каротидная симпатическая денервация). Обращает на себя внимание тот факт, что наиболее важной причиной кардиальных событий, возникающих во время антиадренергического лечения больных с вариантом 1 синдрома удлиненного интервала Q-T, является недостаточная комплаентность [46].
Посредством мультивариантного анализа также были определены предикторы кардиальных событий во время антиадренергической терапии:
удлинение интервала Q-T gt;500 мс;
возникновение жизнеугрожающих аритмий в возрасте до 7 лет;
наличие у пациента вариантов 2 и 3 синдрома.
Носители мутаций KCNH2 или SCN5A имеют относительный риск развития жизнеугрожающих аритмий на фоне приема p-адреноблокаторов, равный 2,81 и 4 соответственно, по сравнению с больными с вариантом 1 синдрома удлиненного интервала Q-T [45]. В соответствии с руководством Американского кардиологического колледжа, Американской ассоциации кардиологов и Европейского общества кардиологов (ACC/AHA/ESC) от 2006 г. [1], использование ИКД у больных с вариантами 2 и 3 (интервал Q-T gt;500 мс) в качестве основного средства профилактики остановки сердца имеет класс доказательности IIB. Поскольку ИКД не предотвращает развитие
аритмий и учитывая молодой возраст пациентов, его имплантация предрасполагает к осложнениям, связанным с разрывом отводящих электродов и ложным срабатыванием. Соответственно поиск терапевтических альтернатив является приоритетным в данной области исследований.
Источник: Кэмм А. Джон, Люшер Томас Ф., Серруис П.В., «Болезни сердца и сосудов.Часть 2 (Главы 6-10)» 2011
А так же в разделе « ГЕНЕТИКА И ПАТОФИЗИОЛОГИЯ »
- РЕЗЮМЕ
- МОНОГЕННЫЕ МЕНДЕЛЕВСКИЕ ЗАБОЛЕВАНИЯ: ОБЩИЙ ОБЗОР
- МОНОГЕННЫЕ ЗАБОЛЕВАНИЯ С ПОРАЖЕНИЕМ МИОКАРДА
- СВЯЗЬ ГЕНОТИП-ФЕНОТИП
- КЛИНИЧЕСКИЙ ПОДХОД К ГЕНЕТИЧЕСКОМУ ТЕСТИРОВАНИЮ
- ГЕНЕТИЧЕСКИЕ ОСНОВЫ И ПАТОФИЗИОЛОГИЯ
- НЕКОМПАКТНЫЙ МИОКАРД ЛЕВОГО ЖЕЛУДОЧКА
- АРИТМОГЕННАЯ КАРДИОМИОПАТИЯ ПРАВОГО ЖЕЛУДОЧКА
- СИНДРОМ МАРФАНА
- ГЕНЕТИЧЕСКИЕ ЗАБОЛЕВАНИЯ БЕЗ ОРГАНИЧЕСКИХ ИЗМЕНЕНИЙ СЕРДЦА
- СИНДРОМ УДЛИНЕННОГО ИНТЕРВАЛА Q-T КЛИНИЧЕСКАЯ КАРТИНА
- ГЕН-СПЕЦИФИЧЕСКИЙ ПОДХОД К ЛЕЧЕНИЮ
- СИНДРОМ БРУГАДА
- ОЦЕНКА РИСКА И ЛЕЧЕНИЕ
- ПРОГРЕССИРУЮЩИМ ДЕФЕКТ ПРОВОДИМОСТИ И СИНДРОМ СЛАБОСТИ СИНУСОВОГО УЗЛА
- ГЕНЕТИЧЕСКИЕ ДЕФЕКТЫ И ПАТОФИЗИОЛОГИЯ
- СИНДРОМ УКОРОЧЕННОГО ИНТЕРВАЛА Q-T КЛИНИЧЕСКАЯ КАРТИНА
- КАТЕХОЛАМИНЕРГИЧЕСКАЯ ПОЛИМОРФНАЯ ЖЕЛУДОЧКОВАЯ ТАХИКАРДИЯ
- ДОПОЛНИТЕЛЬНЫЕ ЛОКУСЫ И ФЕНОКОПИИ
- ГЕНЫ-МОДИФИКАТОРЫ В НАСЛЕДСТВЕННЫХ АРИТМОГЕННЫХНАРУШЕНИЯХ