
В эту группу редко встречающихся геморрагических диатезов входят наследственный дефицит фактора VII (гипопроконвертинемия), фактора X (болезнь Стюарта—Пауэра), фактора II (наследственная гипопротромбинемия) и наследственный комбинированный дефицит всех этих трех факторов, связанный с нарушением их гамма-карбоксилирования в гепатоцитах (см. З.С. Баркаган, 1988).
Диагностика. Клинически при всех этих заболеваниях наблюдается петехи- ально-пятнистый или (при более тяжелых формах) смешанный микроцирку- ляторно-гематомный тип кровоточивости. Лабораторная диагностика базируется на выявлении резко выраженного удлинения протромбинового времени при нормальном тромбиновом времени свертывания, уровне фибриногена в плазме, количестве и функции тромбоцитов. АПТВ остается нормальной при дефиците фактора VII, но удлиняется при дефиците факторов X и II. Добавление к плазме больного нормальной сыворотки крови суточного срока хранения исправляет дефект свертывания (в протромбиновом тесте) при дефиците факторов VII и X, но не корригирует нарушения при дефиците факторов II и V. В коагуляционном тесте с ядом гюрзы нарушение выявляется при дефиците факторов X, II и V, нормальный результат — при дефиците фактора VII, а в тесте с ядом эфы гипокоагуляция обнаруживается только при дефиците фактора II. Дифференциация выявляемых нарушений может проводиться также при помощи фирменных образцов плазм с заведомо известным дефицитом каждого из перечисленных факторов свертывания.
Терапия. При наследственных формах дефицита витамин К-зависимых факторов свертывания лечение препаратами витамина К, в отличие от приобретенных форм данной патологии, остается неэффективным.
В неотложных ситуациях и при отсутствии более действенных препаратов купирование кровотечений осуществляют быстрыми трансфузиями СЗП в дозах не менее 20 мл/кг в сутки. При этом учитывается продолжительность жизни введенных факторов в крови реципиента. Как видно из таблицы 35, наиболее короток этот показатель (всего несколько часов) у фактора VII, в связи с чем его дефицит требует наиболее частых (не менее 3 раз в сутки) и массивных трансфузий плазмы. Полужизнь в кровотоке факторов X и IX занимает промежуточное место и при их дефиците вполне достаточны однократные трансфузии плазмы в сутки. И наконец, очень продолжительна полужизнь в кровотоке фактора II (протромбина), и при его дефиците трансфузии плазмы могут осуществляться один раз в 2—3 дня.
Несравненно более эффективным, особенно при дефиците фактора VII и при глубоком комплексном дефиците нескольких витамин К-зависимых факторов свертывания, является введение концентратов этих факторов — PPSB и др. (см. лечение гемофилии В).
Расчет разовой дозы проводится по формуле:
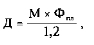
где: Д — доза (ед), М — масса тела больного в кг, — необходимая прибавка уровня ФС в плазме в процентах.
При известном дефиците одного из факторов протромбинового комплекса в терапии могут быть использованы и очищенные концентраты каждого из этих факторов. Практически это может понадобиться лишь для купирования и предупреждения больших кровотечений, связанных с дефицитом наиболее короткоживущего и труднокомпенсируемого фактора VII, хотя применение PPSB в таких ситуациях также достаточно эффективно.
Дефицит фактора У. Этот редко встречающийся геморрагический диатез чаще всего протекает с легкой кровоточивостью, ибо концентрация фактора V в плазме более 15% достаточна для обеспечения хорошего гемостаза. Полу- жизнь этого фактора, введенного в кровоток реципиента, составляет около 35 ч.
Трансфузионная терапия осуществляется СЗП в дозах до 15 мл/кг в сутки. При необходимости повторных трансфузий одновременно назначают мочегонные средства (лазикс, фуросемид).
Дефицит Фактора XIII. Кровоточивостью и плохим заживлением ран дефицит этого фибринстабилизирующего фактора проявляется лишь при его уровне ниже 4% нормы.Распознается эта патология по повторным геморрагиям из плохо заживающих ран (начиная с мокнущего пупка у новорожденных) и быстрого растворения сгустков фибрина из крови больного в 5 М мочевине или 1%-ной монохлоруксусной кислоте.
Лечение состоит из трансфузий СЗП или криопреципитата в дозе до 10—15 мл (ед)/кг один раз в 2—3 дня.
Наследственная гипо(дис! фибриногенемия. Большинство форм аномалий фибриногена протекает бессимптомно, либо с незначительной кровоточивостью, а иногда с наклонностью к тромбозам. Диагностика основывается на выявлении сниженного содержания фибриногена в плазме к замедленному его свертыванию в тромбиновом тесте и (или) в коагуляционных пробах со свертывающими ферментами из змеиных ядов (экарин, ботропклотаза, рептилаза).
Трансфузионная терапия в подавляющем большинстве случаев не нужна. В редких ситуациях компенсация свертывания может быть достигнута трансфузиями СЗП или введениями криопреципитата в дозах до 10 мл (ед)/кг один раз в 2—3 дня. Нет необходимости в более частых введениях этих препаратов, так как период полужизни фибриногена в циркуляции очень велика (см. табл. 35).
Диагностика. Клинически при всех этих заболеваниях наблюдается петехи- ально-пятнистый или (при более тяжелых формах) смешанный микроцирку- ляторно-гематомный тип кровоточивости. Лабораторная диагностика базируется на выявлении резко выраженного удлинения протромбинового времени при нормальном тромбиновом времени свертывания, уровне фибриногена в плазме, количестве и функции тромбоцитов. АПТВ остается нормальной при дефиците фактора VII, но удлиняется при дефиците факторов X и II. Добавление к плазме больного нормальной сыворотки крови суточного срока хранения исправляет дефект свертывания (в протромбиновом тесте) при дефиците факторов VII и X, но не корригирует нарушения при дефиците факторов II и V. В коагуляционном тесте с ядом гюрзы нарушение выявляется при дефиците факторов X, II и V, нормальный результат — при дефиците фактора VII, а в тесте с ядом эфы гипокоагуляция обнаруживается только при дефиците фактора II. Дифференциация выявляемых нарушений может проводиться также при помощи фирменных образцов плазм с заведомо известным дефицитом каждого из перечисленных факторов свертывания.
Терапия. При наследственных формах дефицита витамин К-зависимых факторов свертывания лечение препаратами витамина К, в отличие от приобретенных форм данной патологии, остается неэффективным.
В неотложных ситуациях и при отсутствии более действенных препаратов купирование кровотечений осуществляют быстрыми трансфузиями СЗП в дозах не менее 20 мл/кг в сутки. При этом учитывается продолжительность жизни введенных факторов в крови реципиента. Как видно из таблицы 35, наиболее короток этот показатель (всего несколько часов) у фактора VII, в связи с чем его дефицит требует наиболее частых (не менее 3 раз в сутки) и массивных трансфузий плазмы. Полужизнь в кровотоке факторов X и IX занимает промежуточное место и при их дефиците вполне достаточны однократные трансфузии плазмы в сутки. И наконец, очень продолжительна полужизнь в кровотоке фактора II (протромбина), и при его дефиците трансфузии плазмы могут осуществляться один раз в 2—3 дня.
Несравненно более эффективным, особенно при дефиците фактора VII и при глубоком комплексном дефиците нескольких витамин К-зависимых факторов свертывания, является введение концентратов этих факторов — PPSB и др. (см. лечение гемофилии В).
Расчет разовой дозы проводится по формуле:
где: Д — доза (ед), М — масса тела больного в кг, — необходимая прибавка уровня ФС в плазме в процентах.
При известном дефиците одного из факторов протромбинового комплекса в терапии могут быть использованы и очищенные концентраты каждого из этих факторов. Практически это может понадобиться лишь для купирования и предупреждения больших кровотечений, связанных с дефицитом наиболее короткоживущего и труднокомпенсируемого фактора VII, хотя применение PPSB в таких ситуациях также достаточно эффективно.
Дефицит фактора У. Этот редко встречающийся геморрагический диатез чаще всего протекает с легкой кровоточивостью, ибо концентрация фактора V в плазме более 15% достаточна для обеспечения хорошего гемостаза. Полу- жизнь этого фактора, введенного в кровоток реципиента, составляет около 35 ч.
Трансфузионная терапия осуществляется СЗП в дозах до 15 мл/кг в сутки. При необходимости повторных трансфузий одновременно назначают мочегонные средства (лазикс, фуросемид).
Дефицит Фактора XIII. Кровоточивостью и плохим заживлением ран дефицит этого фибринстабилизирующего фактора проявляется лишь при его уровне ниже 4% нормы.Распознается эта патология по повторным геморрагиям из плохо заживающих ран (начиная с мокнущего пупка у новорожденных) и быстрого растворения сгустков фибрина из крови больного в 5 М мочевине или 1%-ной монохлоруксусной кислоте.
Лечение состоит из трансфузий СЗП или криопреципитата в дозе до 10—15 мл (ед)/кг один раз в 2—3 дня.
Наследственная гипо(дис! фибриногенемия. Большинство форм аномалий фибриногена протекает бессимптомно, либо с незначительной кровоточивостью, а иногда с наклонностью к тромбозам. Диагностика основывается на выявлении сниженного содержания фибриногена в плазме к замедленному его свертыванию в тромбиновом тесте и (или) в коагуляционных пробах со свертывающими ферментами из змеиных ядов (экарин, ботропклотаза, рептилаза).
Трансфузионная терапия в подавляющем большинстве случаев не нужна. В редких ситуациях компенсация свертывания может быть достигнута трансфузиями СЗП или введениями криопреципитата в дозах до 10 мл (ед)/кг один раз в 2—3 дня. Нет необходимости в более частых введениях этих препаратов, так как период полужизни фибриногена в циркуляции очень велика (см. табл. 35).