
- Состояние БЦО при ССВО отличается 28% гипо- биотией, характеризующейся снижением КПп в среднем на 34% и КПд на 24% от адаптивного уровня. Следовательно, ССВО сопровождается таким снижением энергопроизводсгва, которое, хотя и меньше реального ресурсообеспечен ия в среднем на 10%, составляет только 2/з от величины энергопродукции, необходимой для адаптивного ответа организма на микробную нагрузку. Бионеустойчивосгь оказывается обусловленной, прежде всего, перегрузкой иммунной системы, ибо избыточная антигенная нагрузка выводит из строя системы неспецифической защиты (фагоцитарная и система комплемента). В процессе фагоцитоза чужеродных микроорганизмов происходит лизис бактерий, приводящий к образованию бактериальных фрагментов (эндотоксин, пептидогликан-тейхоевые кислоты). Острое несоответствие возможностей фагоцитарной системы микробной нагрузке влечет за собой значительные нарушения функций клеточного и гуморального иммунитета. Это вызывает катастрофическое снижение способности организма сопротивляться инфекции, ответственной за нарушения биоустойчивости и развитие септического процесса.
|
4.1. Бионеустойчивость при ССВО ^ |
|
|
Таблица 4.1 Проявления и медиаторы ССВО |
|
|
Реакция |
Медиаторы |
|
Расширение сосудов |
Фактор Хагемана, брадикинин, лактат, оксид азота, простацгаслин. |
|
Сужение сосудов |
Тромбоксан, лейкотриены В4, С5а |
|
Проницаемость сосудов |
PgE2, СЗа, С5а, лейкотриены С и D, фактор Хагемана, брадикинин, простациклин |
|
Лихорадка |
TNF, IL-1, PgE2 |
|
Хемотаксис полиморфноядерных лейкоцитов, адгезия, фагоцитоз |
IL-1, СЗа, С5а, TNF, фибронекгин, гепатин, лейкотриены, "осколки коллагена", ламинин |
|
Боль |
PgE2 |
- Инициация ССВО эндотоксином играет ключевую роль в развитии АС и септического шока. Проявления и медиаторы ССВО представлены в табл. 4.1.
Эндотоксин выступает в качестве одного из наиболее сильных стимуляторов воспаления в организме человека. Он запускает синтез и секрецию разнообразных эндогенных медиаторов воспаления, включая TNF, IL-I, PAF, эйкозаноиды, опиоиды, а также активирует систему комплемента, свертывающую систему крови, систему контактной активации и др.
Таблица 4.2
Повреждающие эффекты эндотоксина
о Высвобождение эндотоксина при тяжелой инфекции, обусловленной грам (-) флорой, может вызвать системную воспалительную реакцию и распространенное повреждение эндотелия.
о Эндотоксин активирует ряд биологических систем, которые вовлекаются в развитие септического шока:
- кининовую систему;
- систему комплимента;
- систему плазминогена;
- макрофаги, моноциты и др. лейкоциты.
4 Под влиянием эндотоксина происходит активация макрофа
гов/моноцитов, которые продуцируют TNF- . Считается, что именно этот цитокин выполняет функцию основного медиатора септического шока. Внугрибрыжеечное введение высоких доз эндотоксина свиньям приводило к быстрой гибели 50% животных. Процесс танатогенеза сопровождался образованием TNF- . При чём в этот небольшой интервал времени не удалось зарегистрировать изменение концентрации других цитокинов. Введение антагонистов рецепторов PAF (типа BN-52021) подавляло секрецию других медиаторов, образующихся в более позднюю фазу септического шока, что сопровождалось исчезновением некоторых симптомов шока. Однако подобные антагонисты не влияли на образование TNF- . Эти данные позволяют предположить, что именно эндотоксин играет ведущую роль при тяжелых формах септического шока, заканчивающихся гибелью. Основные повреждающие эффекты его приведены в табл. 4.2.
На модели крыс было установлено, что TNF-a достигал максимальной концентрации в плазме уже через 2 ч после в/в введения эндотоксина. Изменений концентраций других цитокинов в этот период времени не наблюдалось. Предварительное введение антагониста BN-50739 не влияло на образование TNF- а. Данные результаты могут объяснить, почему некоторые пациенты устойчивы к интенсивному лечению, направленному против более поздних («вторичных») цитокинов и почему раннее введение анти-TNF-a антител в этих случаях дает положительный защитный эффект.
Аналогично TNF- , IL-1 также играет ведущую роль в воспалительном ответе организма. IL-1 запускает и усиливает ряд процессов во время сепсиса, что приводит к застою крови, увеличению проницаемости сосудов, изменению свертывания крови, клеточной инфильтрации, а также заставляет эндотелиальные клетки продуцировать PGb и PGEi.
В ответ на введение эндотоксина свиньям и крысам происхо- 4
дит секреция PAF. Тесная связь, обнаруженная между тяжестью шока, вызываемого эндотоксином, и секрецией PAF и TNF- , позволяет предположить, что PAF также играет одну из ведущих ролей при септическом шоке. Когда животные выживали в наиболее критические первые 30 минут после инъекции эндотоксина, выход PAF по времени предшествовал продукции TNF- .
В отличие от 2-часового периода времени, когда, при введении эндотоксина крысам, наблюдался максимум концентрации TNF- , при культивировании перитонеальных макрофагов крыс с эндотоксином максимальная концентрация TNF- была зарегистрирована через 24 часа. Таким образом, цитокины по времени образования в ответ на введение эндотоксина условно можно разделить на две группы: ранние и более поздние.
Ранние полипотентные цитокины, например TNF- , IL-1 и PAF, которые высвобождаются в процессе повреждения клеток и тканей, вызывают нарушение гемодинамики и гемодинамическую нестабильность — основные компоненты бионеустойчивости.
Более поздно образующиеся цитокины (IL-6, IL-8, IL-9 и IL-2) оказывают иммуномодуляторное воздействие, приводят к разнообразным эффектам, наблюдаемым при острой фазе сепсиса, в том числе и возоактивным процессам.
Образование PAF, обнаруженное в опытах на свиньях, было тесно связано с секрецией ТхВг, б-кето-PGFia и Шк Раннее увеличение при септическом шоке концентрация ТхВг наблюдалось у свиней, кроликов, собак, крыс. Затем происходило возрастание в плазме концентрации б-кето-PGFia, а выход ITB4 задерживался. Индометацин подавлял продукцию всех эйкоза- ноидов, образующихся циклооксигеназной системой и усиливал выход липоксигеназных продуктов. Освобождение 6-кето- PGFia было мало чувствительно к воздействию индометацина по сравнению с ТхВг. Антагонисты рецепторов PAF эффектив-
но блокировали продукцию ТхЕЬ и незначительно снижали секрецию 6-кето-PGFia.
- Структура и биологическая активность эндотоксина, а также механизмы его взаимодействия с клетками, приводящие к секреции цитокинов, заслуживают более подробного рассмотрения.
Эндотоксин по своей химической структуре является липо- полисахаридом (LPS), встроенным в наружный липидный бислой клеточной стенки грамотрицательных бактерий (рис. 4.1).
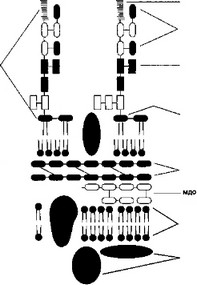
Молекулу эндотоксина условно можно разделить на три час- 4
ти: О-антиген — специфическая цепочка сахаров, центральная часть — Core и липид А.
О-антиген определяет основные иммунные свойства эндотоксина. Благодаря разнообразию этой части молекулы эндотоксина производят иммунологическое типирование бактерий.
С О-антигеном происходит взаимодействие антител человека, что приводит к нейтрализации активности эндотоксина и его последующей элиминации из организма в результате связывания образовавшегося комплекса эндотоксин-антитело с рецепторами к Fc области антитела на поверхности фагоцитирующих клеток. Core связывает между собой О-антиген и липид А. По химическому составу Core представлен гетеро-олигосахаридами. KDO (З-дезокси-О-манно-октулозоновая кислота) и гепто- за образуют внутреннюю область Core эндотоксина, а другие сахара входят в состав наружной области Core.
Наиболее консервативной частью молекулы эндотоксина является ацилированный глюкозаминовый дисахарид — липид А Именно с липидом А связаны токсические свойства эндотоксина. Следует отметить, что ни эндотоксин, ни его липид А клетки человека не убивают. Вызываемые ими токсические эффекты обусловлены образованием клетками человека в ответ на действие LPS (экзогенный агент) разнообразных цитокинов (эндогенные медиаторы). Липид А, подобно эндотоксину, стимулирует образование и секрецию, главным образом, макрофагами/моноцитами TNF- , IL-1, PAF и других цитокинов. Липид А способен непосредственно активировать классический путь активации комплемента в отсутствие антител.
Основные принципы, на которых основаны различные виды биологической активности LPS, в настоящее время хорошо известны. В отличие от действия многих белковых токсинов (токсин ботулизма, дифтерийный токсин и др.), токсические эф-
4 фекты эндотоксинов не связаны с необратимым подавлением различных клеточных функций, которые ведут к гибели клеток и некрозу тканей. Напротив, для действия эндотоксинов требуется активный ответ клеток организма. В соответствии с современными взглядами, LPS через липид А взаимодействует с разнообразными клетками человека, в том числе:
«с макрофагами и моноцитами;
° эндотелиальными клегками; о гладкомышечными клетками;
° полиморфоядерными гранулоцитами;
«тромбоцитами.
Для действия эндотоксинов на биоусгойчивость организма человека и животных особенно важное значение имеют макрофаги/моноциты. Активация макрофагов LPS приводит к продукции разнообразных биологически активных соединений, в том числе производных арахидоновой кислоты (простаглан- дины, тромбоксаны, лейкотриены), оксида азота, активных форм кислорода, пептидных медиаторов, таких, как TNF- , интерлейкины (IL-1, IL-6, IL-8, IL-10), фактор активации тромбоцитов (PAF) и др. Эти вторичные гормоноподобные белки обладают выраженной биологической активностью. При введении в организм они вызывают эффекты, имитирующие действие эндотоксина.
В последние годы стало очевидным, что многие процессы, такие, как индукция устойчивости к инфекциям, адъювантная активность и др., вызываются низкими, физиологическими концентрациями медиаторов. Высокая температура, падение артериального давления, эндотоксиновый шок проявляется при достижении «высоких» концентраций медиаторов в крови и тканях.
Следует, однако, отметить, что низкие концентрации медиаторов способны приводить к возникновению патологических процессов в организме человека на фоне действия LPS. Такое
потенцирование может также провоцироваться и другими при- 4 чинами, в том числе хроническими инфекциями, опухолями, экзотоксинами бактерий. Одними из факторов, повышающих чувствительность к LPS, является г-интерферон.
Ряд факторов позволяет предположить, что на поверхности клеток-мишеней существуют специфические рецепторы, ответственные за взаимодействие с эндотоксином:
о зависимость связывания LPS с макрофагами от концентрации эндотоксина имеет вид кривой с насыщением;
освязывание LPS с клетками конкурентно ингибируется различными производными LPS, в результате чего биологическая активность эндотоксина не проявляется;
°обнаружены мутантные клетки, не отвечающие на эндотоксин.
Эти результаты свидетельствуют о том, что связывание LPS осуществляется специфическими молекулами, расположенными на поверхности клетки (рис. 4.2).
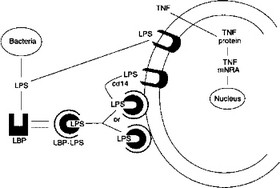
Рис. 4.2. Связывание эндотоксина и активация макрофагов липополисахаридом CD14 — рецепторный белок, связывающий комплекс LBP-LPS;
LBP — сывороточный белок, связывающий LPS;
TNF — фактор некроза опухолей.
4 Более подробные исследования с различными производны
ми липида А эндотоксина позволили установить, что именно эта часть молекулы специфически взаимодействует с рецепторами плазматической мембраны макрофагов/моноцитов. В результате такого взаимодействия происходят синтез и секреция биологически активных цитокинов.
Некоторые не токсичные синтетические производные липида А конкурируют с LPS и липидом А за связывание с рецепторами, выступая в качестве антагонистов эндотоксина. В результате действия на клетки подобных антагонистов подавляется образованием TNF- и других медиаторов.
- Роль в развитии биоустойчивости АНЦА (анти- нейтрофильных цитоплазматических антител) достаточно важна. Они не только являются серологическими маркерами системных васкулитов, но и участвуют в патогенезе ССВО. Установлено, что цитоплазматические ферменты при определенных условиях могут экспонироваться на мембране нейтрофилов, в результате чего они становятся доступными для взаимодействия с АНЦА Обсуждается два принципиально возможных механизма этого феномена. Мембранная экспрессия цитоплазматических ферментов, возможно, имеет место в результате пре- дактивации нейтрофилов провоспалительными цитокинами, такими, как TNF- и IL-8. Полагают, что транслокация и высвобождение цитоплазматических ферментов являются компонентом физиологического ответа нейтрофилов на воспаление, но в некоторых случаях это может быть стимулом для синтеза патогенных АНЦА Более того, получены результаты, свидетельствующие о том, что транслокация компонентов первичных гранул на мембране нейтрофилов может являться результатом не только их цитокинопосредованной предакгивации, но и ускоренного апоп- тоза нейтрофилов. В связи с этим важное значение имеют данные
о том, что для различных аутоиммунных заболеваний характер- 4 ны: ускоренный апоптоз мононуклеарных клеток in vitro и увеличение экспрессии маркеров апоптоза (Fas) АРО-1 и bcl-2 в циркулирующих клетках.
По данным экспериментальных исследований, связывание АНЦА с мембраноассоциированными ферментами вызывает ряд косгимуляторных эффектов, влияющих на функциональную активность нейтрофилов:
о усиление дегрануляции и респираторного взрыва, приводящего к повреждению эндотелиальных клеток (ЭК); о потенцирование нейтрофильного хемотаксиса; о продукция окиси азота;
о усиление адгезии нейтрофилов к культивированным ЭК посредством усиления экспрессии молекул адгезии (ICAM-1, Е-селектин, VCAM-1) на мембране нейтрофилов и ЭК;
о синтез IL-8 ЭК, который обеспечивает прилипание лейкоцитов и лимфоцитов к эндотелию и трансэндотелиальную миграцию этих клеток в ткани;
о увеличение продукции тканевого фактора и тромбомоду- лина, вызывающих развитие гиперкоагуляции на фоне сосудистого воспаления.
Принципиально важное значение имеет тот факт, что активация нейтрофилов АНЦА зависит не только от специфического распознавания антигена, но и от Fc-завиеимого перекрестного связывания АНЦА FcyRIIIB и особенно FcyRIIIB нейтрофилов. Блокирование Fc-рецепторов нейтрофилов соответствующими моноклональными антителами или ингибиторами метал- лопротеиназ подавляет активацию нейтрофилов, вызванную АНЦА Высказана интересная гипотеза, что различная чувствительность к развитию АНЦА-ассоциированных некротизирующих васкулитов может быть частично обусловлена полиморфизмом Fc-рецепторов в отношении авидности связывания с различны-
4 ми субклассами IgG. Совсем недавно было показано, что среди больных гранулематозом Вегенера поражение почек наблюдается достоверно чаще у носителей NAI-аллеля FcyRIIIB. Таким образом, АНЦА-опосредованная активация нейтрофилов может носить как антигенспецифический, так и антигеннеспеци- фический характер, а указанный идиотип может являться маркером «патогенной» популяции АНЦА.
Значение самих нейтрофильных ферментов в иммунопатогенезе бионеустойчивости при ССВО очевидно. Экспериментальные исследования свидетельствуют об их потенциальной деструктивной активности. Например, перфузия протеина ПР-3 лабораторным животным вызывает развитие эмфиземы, а иммунизация миелопероксидазой индуцирует развитие нефрита. ПР-3 и эластаза вызывают отслойку и лизис ЭК. Кроме того, ПР-3 обладает способностью расщеплять и инактивировать CI-ингибитор, что ассоциируется с усилением комплементза- висимого повреждения тканей. Однако увеличение уровня ПР-3 в сыворотке выявлено не только у больных с АНЦА-ассо- циированными васкулитами (гранулематоз Вегенера), но и при системной красной волчанке (СКВ). При иммуноморфологи- ческом исследовании ПР-3 и эластаза идентифицированы в почках больных при быстропрогрессирующем нефрите, как связанном с АНЦА, так и не ассоциирующемся с гиперпродукцией этих антител. Данные, касающиеся влияния АНЦА на активность циркулирующих нейтрофильных ферментов, противоречивы. С одной стороны, АНЦА обладают способностью ингибировать ферментативную активность ПР-3 за счет связывания с каталитическим доменом фермента и предотвращают отслойку и лизис ЭК, индуцированную ПР-3 in vitro. С другой стороны, связывание АНЦА с ПР-3 отменяет инактивацию ПР-
- ингибитором а 1 -антитрипсина и потенцирует деструктивный эффект этого фермента.
Другой важный патогенетический механизм АНЦА-ассоци- 4 ированных васкулитов может бьггь обусловлен перекрестным взаимодействием АНЦА с сосудистым эндотелием. Имеются данные о том, что ПР-3 и миелопероксидаза, высвобождаясь из нейтрофилов в процессе их активации, связываются с мембраной ЭК или могут даже непосредственно синтезироваться и транслоцироваться на мембране ЭК при активации эндотелия цитокинами (IL-1, TNF- и г-интерферон). Присутствие нейт- рофильных ферментов на мембране ЭК является строго доказанным и, несомненно, служит одной из важных причин перекрестной реактивности АНЦА с ЭК. В других исследованиях обнаружена перекрестная реактивность АНЦА с лейкоцитарным (gp 170/80-110) и эндотелиальным (gp 130) мембранными гликопротеинами. Примечательно, что gp 170/80-110 оказался идентичным лейкоцитарному белку h-lamp-2, которому придается существенное значение в метасгазировании опухолей и развитии тканевого фиброза. Показано также, что активация лейкоцитов приводит к гиперэкспрессии gp 170/80-110 на мембране этих клеток, причем 1ашр-ассоциированные гликопротеины выполняют функцию рецепторов для селектинов.
Все это вместе взятое свидетельствует о том, что АНЦА способны вызывать активацию нейтрофилов, а также обладают ан- тиэндотелиальной активностью, что, вероятно, и определяет патогенетический потенциал этих антител при ССВО.
- Антиэндотелиальные клеточные антитела (АЭКА), обнаруженные при ряде аутоиммунных и хронических воспалительных заболеваний, могут рассматриваться в качестве возможного патогенетического фактора сосудистой патологии при ССВО и различных заболеваниях человека. В некоторых случаях АЭКА могут повреждать ЭК посредством комплементзависи- мого цитолиза или антителозависимой клеточной цитотоксич-
4 ности. Описаны две популяции антител, одна из которых лизи- рует клетки эндотелия, обработанные IF- , а другая — клетки, прединкубированные с IL-1 и TNF- . Наряду с цитотксич- ностью, патогенный потенциал АЭКА может реализоваться за счет изменения функциональной активности эндотелия. Так, по данным N. Del Papa и соавт., АЭКА индуцируют экспрессию молекул адгезии (Е-селектина и VCAM-1, ICAM-1) на мембране ЭК и синтез провоспалительных цитоктинов (ИЛ-6, ИЛ-8 и мо- ноцитарного хемотактического белка 1).
- Антифосфолипидные антитела (АФЛ) являются маркерами эндотелиального повреждения и могут выступать в качестве важного дополнительного патогенетического механизма тромботических процессов, нередко осложняющих течение ССВО.
- Цитотоксические Т-лимфоциты могут повреждать клетки сосудистого эндотелия, распознавая аллогенные антигены главного комплекса гистосовместимости (ГКГ), экспрессирующие на их мембране. Этот механизм может иметь особое значение при ССВО. Кроме того, по данным экспериментальных исследований, CD8+ Т-лимфоциты, распознающие гетеро- логичные антигены класса 1 ГКГ, принимают участие в поражении сосудистой стенки, индуцированном переносом чужеродных генов ГКГ в стенку артерий. Цитотоксические лимфоциты ( / Т-лимфоциты), секретирующие перфорин, выявлены в стенке сосудов. Интересно, что в зонах, инфильтрированных этими лимфоцитами, отмечается экспрессия стрессорного белка. Эти данные позволяют предположить, что поражение сосудистой стенки при ССВО может бьггь связано с двумя типами цитотоксических реакций. Во-первых, с клеточной цитотоксичностью, опосредуемой С08-лимфоцитами, экспрессирующими
/ -рецепторы, которая рестриктирована по классу 1 ГКГ; во^ 4 вторых, с цитотоксичносгью Т-лимфоцитов, экспрессирующих / Т-КР, распознающих стрессорные белки. Гиперэкспрессия определенных типов V генных сегментов Т-КР была обнаружена на небольшой части клонированных CD4+ Т-лимфоцитов. Предполагается, что это связано с клональной пролиферацией субпопуляции Т-лимфоцитов, распознающих антиген, присутствующий в стенке сосуда.
- Гиперэкспрессия молекул адгезии и соответствующих лейкоцитарных лигандов имеет значение в развитии бионеустойчивости при ССВО. Индукция адгезина может быть опосредована взаимодействием АНЦА и АЭКА с поверхностью ЭК.
Кроме того, экспрессия этих молекул имеет важное значение в иммунокомплексном и комплементзависимом повреждении сосудистой стенки. Продукты активации комплемента (например, CI) индуцируют экспрессию Е-селектина, ICAM-1 и VCAM-1 в культуре ЭК. Примечательно, что эстрогены, играющие роль в патогенезе аутоиммунных ревматических заболеваний и некоторых форм системных васкулитов (артериит Такая- си), потенцируют TNF- -зависимую гиперэкспресию молекул адгезии на сосудистом эндотелии.
- Накопленные к настоящему времени фундаментальные данные по патофизиологии острого воспалительного ответа (кстати, во многом благодаря разработке антимедиатор- ной терапии) позволили отметить, что эволюция ССВО сопровождается изменениями регуляции экспрессии определенных генов. Таким образом, медиаторно-цитокиновую реакцию можно представить и как следствие дисбаланса генетических механизмов рефляции медиаторного ответа при ССВО, что может вносить качественные преобразования в работу метаболического
- аппарата в ответ на изменения условий среды. В самом общем виде скорость метаболических реакций определяется индукцией или репрессией синтеза всех ферментов соответствующего метаболического пути. Механизмы индукции и репрессии предохраняют клетку от напрасной траты аминокислот и энергии и определяются экспрессией тех или иных генов. Известно, что в основе механизмов регуляции транскрипция (считывания) гена лежит принцип оперонной организации ДНК, во многом разработанный благодаря изучению генетики бактерий (Е. coli), когда один из нескольких генов, кодирующих аминокислотные последовательности белков, вместе с прилегающими регуляторными участками ДНК образуют элементарную единицу транскрипции — оперон. Имеется по крайней мере четыре компонента системы: структурный ген, ген-регулятор, оператор и промотор. Структурные гены, как правило, располагаются кластерно (группами). 1ен-регулятор определяет структуру бел- ка-репрессора. Этот белок способен связываться с оператором, который контролирует функционирование прилежащего кластера структурных генов. Промотор — участок ДНК, выполняющий роль сайта связывания с РНК-полимеразой, ферментом, катализирующим синтез шРНК с нуклеотидной последовательностью ДНК. Если белок-репрессор связан с оператором, то РНК-полимераза не может присоединиться к промотору и/или перемещаться, транскрибируя РНК структурного гена, т.е., соответствующий фермент не синтезируется. Когда оператор свободен от белка-репрессора, РНК-полимераза, присоединившись к промотору, может транскрибировать ДНК. Репрессор представляет собой аллосгерический белок, содержащий как минимум 2 центра. Один обладает сродством к нуклеотидной последовательности оператора, а другой — к молекуле специфического индуктора. Присоединение индуктора к репрессору изменяет его конформацию, снижая тем самым сродство первого
аллостерического центра к оператору. Индуктор необязательно 4 вещество белковой природы, это может быть и низкомолекулярный продукт. Следует отметать, что объединение генов, ответственных за образование ферментов какого либо метаболического пути, не является непременным условием для регуляции их функции специфическим белком-репрессором. Одним и тем же белком могут контролироваться несколько промоторов с прилегающими структурными генами, расположенными на различных участках хромосомы (для прокариот) и/или различных хромосомах (для эукариот). Например, совокупность генов, кодирующих около двух десятков белков и ферментов, которые инициируют у бактерий ответ на воздействие, повреждающее ДНК, так называемый SOS —ответ, регулируются одним репрес- сором — продуктом гена 1ехА.
Для высших организмов, в том числе и человека, в регуляции транскрипции необходимо участие по крайней мере еще одного гена — гена сенсора, предположительно модулируемого гормонами. Имеется 3 различные РНК-полимеразы, а наличие на ДНК не транслируемых участков (интронов) требует в последующем механизма созревания (сплайнсинга) тРНК.
Следующим принципиальным моментом, лимитирующим выход конечного продукта, является трансляция тРНК. Выход белка при трансляции определяется как количеством копий тРНК, так и числом функционирующих рибосом. Доза рибо- сомных генов генетически детерминирована для каждого конкретного индивидуума.
Липополисахарид оказывает стимулирующее влияние на многие пути внутриклеточной активации. Важным участком этих процессов является рецептор CD14 плазматической мембраны клеток, который взаимодействует с комплексом, состоящим из эндотоксина и липопсшисахарид-связывающего белка плазмы. В результате активируется ядерный фактор транскрип-
- ции NF-kB, который в неактивной форме связан цитозольным белком Ik-BA. Происходит индуцируемое фосфорилирование белка Ik-BA, что показано на рис. 4.3.
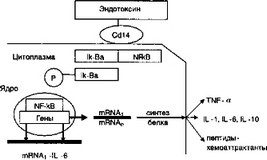
Рис. 4.3. Передача сигнала от эндотоксина на геном
Далее фактор транскрипции NF-kB связывается с регуляторными генами провоспалительных цитокинов и происходит индукция синтеза цитокинов (TNF- , IL-1, IL-6, IL- 10 и др.) и пептидов-хемоаттрактантов для нейтрофилов и моноцитов.
Важно отметить способность эндотоксина к агломерации или опсонизации клеток организма, в первую очередь эндотелиальных. Необходимое условие такого процесса — высокая концентрация эндотоксина в сыворотке крови. Это неспецифическое связывание с клетками эндотелия при активации комплемента, клеток крови (в первую очередь макрофагов и нейтрофилов) и гемокоагуляции может приводить к обширным повреждениям эндотелия, что является краеугольным камнем в развитии при ССВО полиорганной недостаточности — основной причины летальных исходов при сепсисе.
Несомненно, что в идеале патогенетически обоснованной 4 мишенью антимедиаторной терапии должен стать либо репрес- сор, либо индуктор, способные контролировать транскрипцию основных септических медиаторов.
Имеются сообщения, что индуцибельный контроль генов цитокинов (по крайней мере, TNF), рецепторов цитокинов, главного комплекса антигенов гисгосовместимости, острофазных белков, а по некоторым данным и макрофагальной синтазы оксида азота, осуществляется посредством белка ядерного матрикса — ядерный фактор транскрипции kB/NF-kB. Причем для индукции шРНК цитокинов (TNF, IL-1) требуется активация (фосфорилирование) протеинкиназы-С и протеинтирозинки- назы. Это приводит к протеолитическому расщеплению ингибиторной формы и последующей инициации транскрипции соответствующих генов, т.е. можно представлять NF-kB как реп- рессор при наличии индуктора в виде протеинтирозинкиназы.
Однако необходимо признать, что, как подавляющее большинство принципиально важных аспектов проблемы регуляции экспрессии генов человека, так и методологические подходы к изучению данного вопроса в настоящее время не ясны. Не удивительны поэтому неожиданные, патогенетически не объяснимые находки ранних маркеров генерализации инфекционного процесса, в частности, работы, указывающие на высокую прогностическую значимость изучения уровня поокальиитони- на в крови пациентов с ССВО.
Хотя по-прежнему основной целью патогенетического направления терапии ССВО/ПОН остается контроль медиаторного ответа хозяина, на сегодняшний день нет такого типа терапевтического воздействия, в отношении которого была бы показана его эффективность в контролируемых клинических испытаниях.
Известно, что выброс воспалительных медиаторов достаточно неспецифический процесс. Любой повреждающий агент (ги-
- поксия, аминокислотное голодание, антигенное воздействие, термические факторы и др.) способен привести к медиаторному взрыву, который носит в целом стереотипный характер, в сущности, представляя собой констуитивный ответ организма/клетки на изменение условий существования.
ССВО не есть прерогатива сугубо инфекции. Поэтому не совсем логичным представляется воздействовать непосредственно лишь на медиаторы септического каскада, т.е. практически на ранние последствия повреждающих событий, без коррекции причин патологического стресса. Нет смысла не замечать эффекта от известных и общепринятых методов лечения.
Доказано, что 4 компонента интенсивной терапии реально влияют на выживаемость при инфекционной патологии — ранняя диагностика генерализации инфекционного процесса, рациональная антибактериальная химиотерапия, оптимизация кислородного транспорта, полноценное возмещение калорических потребностей и белка. С учетом комплексного действия медиаторов воспаления, только комбинированная терапия может эффективно контролировать течение ССВО и предотвращать формирование ПОН.
Представленные данные позволяют утверждать, что основным направлением патогенетической профилактики АС является комплекс ИТ, направленный на стабилизацию БЦО при ССВО. При этом реальность современного терапевтического подхода к проблеме ССВО должна заключаться в сдержанном отношении к существующим методам непосредственного антиме- диаторного воздействия, сфокусированности лечения, в том числе и медиаторных расстройств, на уже подтвердивших свою значимость компонентах интенсивной терапии: адекватной антимикробной химиотерапии, полноценной инфузионно-транс- фузионной терапии, своевременной поддержке или протезированию недостаточно функционирующих органов (систем) и БЦО.