АРИТМОГЕННАЯ КАРДИОМИОПАТИЯ
ОПРЕДЕЛЕНИЕ Согласно классификации КМП, принятой ВОЗ в 1996 г., к АКМП относят группу заболеваний миокарда, характеризующихся структурными и функциональными нарушениями ПЖ, возникшими вследствие локальной или диффузной атрофии и замещения миокарда жировой или фиброзной тканью [38]. Чаще всего поражаются выносящий тракт, верхушка и подклапанная область
свободной стенки ПЖ. МЖП в патологический процесс обычно не вовлекается и остается интактной [196]. Наиболее часто АКМП страдают мужчины в возрасте 15-35 лет [197].
Морфологические изменения в сердце приводят к развитию региональной или глобальной дисфункции ПЖ и жизнеугрожающих нарушений ритма, исходящих из ПЖ (ЖЭС, устойчивая ЖТ, ФЖ), что может послужить причиной ВСС у молодых пациентов с предположительно здоровым сердцем[196, 198].
АКМП впервые описана Фонтеном (Fontaine) и соавт. в 1977 г. С тех пор достигнут значительный прогресс в понимании патогенеза, патологической анатомии и клинической картины этого заболевания [199-204].
В то же время все еще отсутствует полная информация о генетической предрасположенности и течении АКМП. Требуют дальнейшего изучения и разработки способы диагностики, стратификация риска, а также методы лечения пациентов с высоким риском развития заболевания.
ЭТИОЛОГИЯ И РАСПРОСТРАНЕННОСТЬ
Причинами замещения миокарда ПЖ жировой тканью при АКМП могут быть апоптоз кардиомиоцитов [205], воспалительные изменения [196, 202, 206], генетическая предрасположенность к атрофии миокарда [202], а также участие вирусной инфекции в патогенезе АКМП [207]. Известны отдельные регионы в Греции и северной Италии, где семейный анамнез наводит на мысль о наследственном характере заболевания с аутосомно-доминантным типом наследования, различной пенетрантностью и фенотипами [197, 208-210]. В связи с трудностью диагностики АКМП, а также с тем, что у многих больных отсутствуют клинические симптомы и болезнь может дебютировать с ВСС, истинная распространенность АКМП остается неизвестной [211].
ГЕНЕТИКА
Семейный анамнез при АКМП можно проследить в 30-50% случаев [197]. Также возможны спорадические или семейные формы заболевания с неполной пенетрантностью и изменчивым фенотипом. Носителями этого нарушения могут быть как мужчины, так и женщины. Кроме того, и те, и другие могут передавать его по наследству.
В настоящее время обнаружено десять локусов в семи хромосомах, мутации которых могут привести к возникновению АКМП (табл. 18.9) [212]. Были идентифицированы три белка, нарушение выработки которых индуцирует развитие заболевания. В случае редкой наследственной формы АКМП с пенетрантностью 90% установлен дефект в гене хромосомы 17, кодирующем цитоскелетный белок плакоглобин [213]. Генетический анализ при схожей форме АКМП, для которой характерны кератодермия, густые кучерявые волосы в детстве и развитие СН в юности, позволил обнаружить мутацию в хромосоме 6p23-p24, кодирующей десмоплакин, - белок, необходимый для прикрепления промежуточных филаментов к десмосоме [214]. Мутация рианодиновых рецепторов сердца (хромосома 1q23-q24) приводит к уменьшению высвобождения внутриклеточного кальция, возникновению электрической нестабильности и, в результате, к возникновению ЖТ.
Таблица 18.9. Генетические изменения при аритмогенной кардиомиопатии*
Изменено (с разрешения): Paul M., Schulz-Bahr E., Breithard G. et al. Genetics of arrhythmogenic right ventricular cardiomyopathy: status quo and future perspectives // Z. Kardiol. - 2003. - Vol. 92. - P. 128136.
ПАТОФИЗИОЛОГИЯ
Для АКМП характерно приобретенное замещение миокарда переднебоковой стенки ПЖ жировой или фиброзно-жировой тканью. Процесс распростра-няется волнообразно - от эпикарда к эндокарду. В 80% случаев происходит диффузное жировое замещение миокарда, и в 20% - региональное (сегментарное) замещение. Трансмуральная инфильтрация часто ассоциирована с увеличением толщины стенки желудочка. В 50% случаев обнаруживают мешкообразные аневризмы в области верхушки, задненижней стенки или выносящего тракта ПЖ, формирующиеся в результате сочетанного замещения жировой и фиброзной тканью. При гистоморфологическом исследовании находят атрофию миокарда и фокальный некроз кар-диомиоцитов в сочетании с лимфоцитарной инфильтрацией миокарда, сходной с таковой при хроническом миокардите. Воспаление миокарда [206, 215], вирусная инфекция [207], а также генетически детерминированная дистрофия миокарда могут быть вызваны гибелью кардиомиоцитов в результате апоптоза [205, 216]. Участки сохранившегося миокарда, расположенные между жировой или фиброзно-жировой тканью, способны быть потенциальными источниками возникновения возвратной тахикардии.
КЛИНИЧЕСКАЯ КАРТИНА
Наиболее характерный клинический признак АКМП - симптомная ЖТ, возникающая при физической нагрузке и имеющая правожелудочковое происхождение (форма БЛНПГ). Вследствие устойчивой или неустойчивой ЖТ часто возникают сердцебиения и обмороки. Что касается морфологии ЖТ, то иногда сложно дифференцировать нарушения ритма при АКМП и доброкачественную ненаследственную идиопатическую ПЖ-тахикардию, возникающую в области выносящего тракта ПЖ, или реципрокную АВ-тахикардию. У пациентов, не получающих полноценное лечение, ЖТ может трансформироваться в ФЖ. Вследствие того что у многих больных симптомы отсутствуют, и первым признаком болезни может быть ВСС, истинная распространенность желудочковой фибрилляции, связанной с АКМП, остается неизвестной. У пациентов с ХСН и желудочковыми нарушениями ритма в анамнезе или без них АКМП часто остается нераспознанной, так как этих больных считают пациентами с ДКМП.
ДИАГНОСТИКА
Тщательно собранный анамнез, жалобы на сердцебиения, нарушения ритма, головокружения, предобморочные и обморочные состояния, а также семейный анамнез могут облегчить диагностику заболевания.
В 90% случаев при АКМП присутствуют изменения на ЭКГ. Наиболее часто поражение ПЖ ассоциировано с возникновением отрицательного зубца Т в грудных отведениях V1-V3 без БПНПГ (рис. 18.24). Нарушения реполяризации в отведениях, расположенных выше V3, свидетельствуют о вовлечении в патологический процесс ЛЖ [200]. Патогномоничные ЭКГ-признаки АКМП - БПНПГ, удлинение комплекса QRS в отведениях V1-V3 и волна эпсилон (см. рис. 18.24), которая возникает в 30% случаев вследствие поствозбуждения ПЖ и отражает его замедленную деполяризацию. Волна эпсилон может быть атипичной и выглядеть как "гладкий" потенциал, сформированный атипично длинным зубцом R' в отведениях V1-V3, в случае, если с задержкой возбуждается большое количество волокон миокарда. Именно поэтому длительность комплекса QRS в
отведениях V1-V3, превышающую 25 мс от его продолжительности в отведении V6, следует расценивать как волну эпсилон [200].
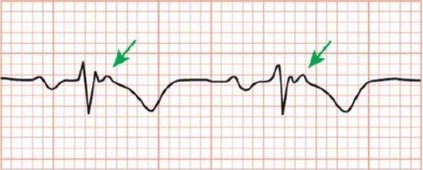
Рис. 18.24. ЭКГ пациента с аритмогенной кардиомиопатией: инверсия зубца Т и эпсилон- волна в отведении V1.
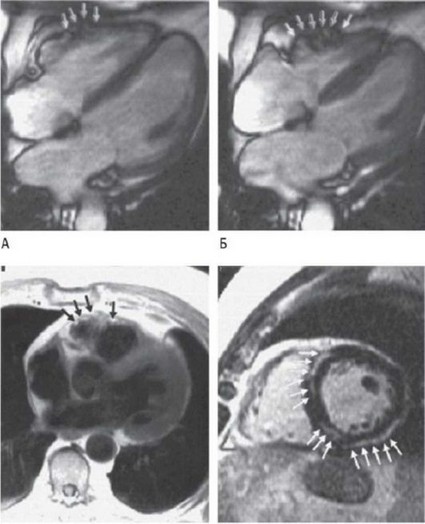
"Золотой стандарт" диагностики АКМП - рентгеноконтрастная вентрикулография ПЖ. Обнаруживают характерную дилатацию ПЖ в сочетании с региональными нарушениями сократительной способности или дискинезией по типу выпячиваний и аневризм в области выходного тракта ПЖ, верхушки или трикуспидального клапана. Эти же признаки, имеющие специфичность 90%, можно обнаружить и с помощью таких неинвазивных методов исследования, как МРТ сердца и ЭхоКГ. МРТ позволяет точно охарактеризовать функцию и анатомию ПЖ, но этот метод имеет ограниченные диагностические возможности. Например, невозможно оценить точный размер утолщения свободной стенки ПЖ и количество жировой ткани по сравнению с ее обычным содержанием в эпикарде и перикарде (рис. 18.25). В настоящее время ЭхоКГ - наиболее ценный метод, поскольку позволяет провести дифференциальную диагностику, исключить другие заболевания сердца, уточнить функциональную способность ПЖ и ЛЖ и обнаружить их структурные аномалии. ЭхоКГ широко применяют для ранней диагностики прогрессирования заболевания, а также для скринингового обследования членов семьи пациента. При малых формах АКМП локализацию структурных изменений ПЖ обнаружить достаточно сложно. К сожалению, незначительные отклонения от нормы можно не диагностировать даже при использовании всех методов исследования.

Обнаружение типичных гистологических изменений in vivo при АКМП с помощью эндомиокардиальной биопсии имеет определенную диагностическую ценность, но ограничено низкой чувствительностью метода (особенно при невыраженных формах АКМП). Эндомиокардиальная биопсия не может подтвердить трансмуральное замещение миокарда жировой или фиброзно-жировой тканью. Более того, фокальные или сегментарные изменения, отсутствие вовлечения в патологический процесс МЖП и естественное присутствие жировой ткани у пожилых пациентов может привести к гипердиагностике заболевания на основании результатов
биопсии. Повышенное содержание жировой или фиброзной ткани у пожилых пациентов или другие признаки КМП также снижают диагностическую ценность МРТ.
На основании клинических и морфологических изменений для АКМП были установлены диагностические критерии, представленные в табл. 18.10 [217]. Диагноз АКМП базируется на обнаружении двух больших критериев, или одного большого и двух малых критериев, или четырех малых критериев, подтвержденных гистологическими, ЭКГ-находками и существованием аритмии [218].
Таблица 18.10. Большие и малые критерии диагностики аритмогенной кардиомиопатии*
Большие критерии
Семейное заболевание, подтвержденное при аутопсии или хирургическом вмешательстве
Существование волны эпсилон или удлинение комплекса QRSgt;110 мс в отведениях V1-V3
Выраженная дилатация ПЖ и систолическая дисфункция с или без незначительного вовлечения в патологический процесс ЛЖ
Локальные аневризматические расширения ПЖ (зоны акинезии или дискинезии с диастолическим выбуханием)
Выраженная сегментарная дилатация ПЖ
Замещение миокарда фиброзно-жировой тканью (по результатам эндомиокардиальной биопсии)
Малые критерии
Указания на ВСС в семейном анамнезе в возрасте до 35 лет
Семейный анамнез заболевания, подтвержденного диагностическими критериями
Поздние потенциалы на сигнал-усредненной ЭКГ
Инверсия зубца Т в отведениях V2 и V3 без БПНПГ у пациентов в возрасте старше 12 лет
Тахикардия с формой комплекса, соответствующей БЛНПГ (устойчивали или неустойчивая) на ЭКГ, при ХМ или во время пробы с физической нагрузкой
Более 1 тыс. ЖЭС в сутки при ХМ
Небольшая дилатация ПЖ с или без его дисфункции на фоне сохраненной функции ЛЖ
Незначительная сегментарная дилатация ПЖ
Участки региональной гипокинезии правых отделов
Изменено (с разрешения): McKenna W.J., Thiene G., Nava A. et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy // Br. Heart J. - 1994. - Vol. 71. - P. 215-218.
СТРАТИФИКАЦИЯ РИСКА
Основная цель лечения АКМП - профилактика возникновения жизнеугрожающих нарушений ритма и ВСС. Вероятность развития стресс-индуцированной ФЖ или гемодинамически значимой ЖТ
прогнозировать сложно, особенно у асимптомных молодых людей и атлетов. В настоящее время отсутствуют рекомендации о назначении профилактического лечения пациентам с установленным диагнозом АКМП, минимальными клиническими симптомами или морфологическими изменениями. Асимптомные пациенты с высоким риском возникновения ВСС имеют отягощенный семейный анамнез, желудочковые нарушения ритма в детском или подростковом возрасте или во время занятий спортом, обмороки или вовлечение в патологический процесс ЛЖ. Прогностическая ценность ЭКГ, ХМ, нагрузочных тестов и сигнал-усредненной ЭКГ в качестве предиктора ВСС невысока. Пациентов с клиническими симптомами необходимо обследовать более тщательно и при необходимости проводить ангиографию ЛЖ, программируемую желудочковую стимуляцию и эндомиокардиальную биопсию.
свободной стенки ПЖ. МЖП в патологический процесс обычно не вовлекается и остается интактной [196]. Наиболее часто АКМП страдают мужчины в возрасте 15-35 лет [197].
Морфологические изменения в сердце приводят к развитию региональной или глобальной дисфункции ПЖ и жизнеугрожающих нарушений ритма, исходящих из ПЖ (ЖЭС, устойчивая ЖТ, ФЖ), что может послужить причиной ВСС у молодых пациентов с предположительно здоровым сердцем[196, 198].
АКМП впервые описана Фонтеном (Fontaine) и соавт. в 1977 г. С тех пор достигнут значительный прогресс в понимании патогенеза, патологической анатомии и клинической картины этого заболевания [199-204].
В то же время все еще отсутствует полная информация о генетической предрасположенности и течении АКМП. Требуют дальнейшего изучения и разработки способы диагностики, стратификация риска, а также методы лечения пациентов с высоким риском развития заболевания.
ЭТИОЛОГИЯ И РАСПРОСТРАНЕННОСТЬ
Причинами замещения миокарда ПЖ жировой тканью при АКМП могут быть апоптоз кардиомиоцитов [205], воспалительные изменения [196, 202, 206], генетическая предрасположенность к атрофии миокарда [202], а также участие вирусной инфекции в патогенезе АКМП [207]. Известны отдельные регионы в Греции и северной Италии, где семейный анамнез наводит на мысль о наследственном характере заболевания с аутосомно-доминантным типом наследования, различной пенетрантностью и фенотипами [197, 208-210]. В связи с трудностью диагностики АКМП, а также с тем, что у многих больных отсутствуют клинические симптомы и болезнь может дебютировать с ВСС, истинная распространенность АКМП остается неизвестной [211].
ГЕНЕТИКА
Семейный анамнез при АКМП можно проследить в 30-50% случаев [197]. Также возможны спорадические или семейные формы заболевания с неполной пенетрантностью и изменчивым фенотипом. Носителями этого нарушения могут быть как мужчины, так и женщины. Кроме того, и те, и другие могут передавать его по наследству.
В настоящее время обнаружено десять локусов в семи хромосомах, мутации которых могут привести к возникновению АКМП (табл. 18.9) [212]. Были идентифицированы три белка, нарушение выработки которых индуцирует развитие заболевания. В случае редкой наследственной формы АКМП с пенетрантностью 90% установлен дефект в гене хромосомы 17, кодирующем цитоскелетный белок плакоглобин [213]. Генетический анализ при схожей форме АКМП, для которой характерны кератодермия, густые кучерявые волосы в детстве и развитие СН в юности, позволил обнаружить мутацию в хромосоме 6p23-p24, кодирующей десмоплакин, - белок, необходимый для прикрепления промежуточных филаментов к десмосоме [214]. Мутация рианодиновых рецепторов сердца (хромосома 1q23-q24) приводит к уменьшению высвобождения внутриклеточного кальция, возникновению электрической нестабильности и, в результате, к возникновению ЖТ.
Таблица 18.9. Генетические изменения при аритмогенной кардиомиопатии*
| Хромосома | Ген | Авторы |
|
Аутосомно-доминантный тип наследования |
||
| 14q23-q24 | - | Рампаццо и др. [208] |
| 1q42-q43 | Рианодиновые рецепторы | Рампаццо и др. [219] |
| 14q12-q22 | - | Северини и др. [220] |
| 2q32.1-q32.3 | - | Рампаццо и др. [221] |
| 3p23 | - | Ахмад и др. [222] |
| 10q22.3 | - | Мельберг и др. [223] |
| 10p12-p14 | - | Ли и др. [224] |
| 6p24 | Десмоплакин | Рампаццо и др. [225] |
|
Аутосомно-рецессивный тип наследования |
||
| 14q24-q | Плакоглобин | Франс и др. [226] |
| 17q21 | Болезнь Наксос | Кунар и др. [213] |
Изменено (с разрешения): Paul M., Schulz-Bahr E., Breithard G. et al. Genetics of arrhythmogenic right ventricular cardiomyopathy: status quo and future perspectives // Z. Kardiol. - 2003. - Vol. 92. - P. 128136.
ПАТОФИЗИОЛОГИЯ
Для АКМП характерно приобретенное замещение миокарда переднебоковой стенки ПЖ жировой или фиброзно-жировой тканью. Процесс распростра-няется волнообразно - от эпикарда к эндокарду. В 80% случаев происходит диффузное жировое замещение миокарда, и в 20% - региональное (сегментарное) замещение. Трансмуральная инфильтрация часто ассоциирована с увеличением толщины стенки желудочка. В 50% случаев обнаруживают мешкообразные аневризмы в области верхушки, задненижней стенки или выносящего тракта ПЖ, формирующиеся в результате сочетанного замещения жировой и фиброзной тканью. При гистоморфологическом исследовании находят атрофию миокарда и фокальный некроз кар-диомиоцитов в сочетании с лимфоцитарной инфильтрацией миокарда, сходной с таковой при хроническом миокардите. Воспаление миокарда [206, 215], вирусная инфекция [207], а также генетически детерминированная дистрофия миокарда могут быть вызваны гибелью кардиомиоцитов в результате апоптоза [205, 216]. Участки сохранившегося миокарда, расположенные между жировой или фиброзно-жировой тканью, способны быть потенциальными источниками возникновения возвратной тахикардии.
КЛИНИЧЕСКАЯ КАРТИНА
Наиболее характерный клинический признак АКМП - симптомная ЖТ, возникающая при физической нагрузке и имеющая правожелудочковое происхождение (форма БЛНПГ). Вследствие устойчивой или неустойчивой ЖТ часто возникают сердцебиения и обмороки. Что касается морфологии ЖТ, то иногда сложно дифференцировать нарушения ритма при АКМП и доброкачественную ненаследственную идиопатическую ПЖ-тахикардию, возникающую в области выносящего тракта ПЖ, или реципрокную АВ-тахикардию. У пациентов, не получающих полноценное лечение, ЖТ может трансформироваться в ФЖ. Вследствие того что у многих больных симптомы отсутствуют, и первым признаком болезни может быть ВСС, истинная распространенность желудочковой фибрилляции, связанной с АКМП, остается неизвестной. У пациентов с ХСН и желудочковыми нарушениями ритма в анамнезе или без них АКМП часто остается нераспознанной, так как этих больных считают пациентами с ДКМП.
ДИАГНОСТИКА
Тщательно собранный анамнез, жалобы на сердцебиения, нарушения ритма, головокружения, предобморочные и обморочные состояния, а также семейный анамнез могут облегчить диагностику заболевания.
В 90% случаев при АКМП присутствуют изменения на ЭКГ. Наиболее часто поражение ПЖ ассоциировано с возникновением отрицательного зубца Т в грудных отведениях V1-V3 без БПНПГ (рис. 18.24). Нарушения реполяризации в отведениях, расположенных выше V3, свидетельствуют о вовлечении в патологический процесс ЛЖ [200]. Патогномоничные ЭКГ-признаки АКМП - БПНПГ, удлинение комплекса QRS в отведениях V1-V3 и волна эпсилон (см. рис. 18.24), которая возникает в 30% случаев вследствие поствозбуждения ПЖ и отражает его замедленную деполяризацию. Волна эпсилон может быть атипичной и выглядеть как "гладкий" потенциал, сформированный атипично длинным зубцом R' в отведениях V1-V3, в случае, если с задержкой возбуждается большое количество волокон миокарда. Именно поэтому длительность комплекса QRS в
отведениях V1-V3, превышающую 25 мс от его продолжительности в отведении V6, следует расценивать как волну эпсилон [200].
Рис. 18.24. ЭКГ пациента с аритмогенной кардиомиопатией: инверсия зубца Т и эпсилон- волна в отведении V1.
"Золотой стандарт" диагностики АКМП - рентгеноконтрастная вентрикулография ПЖ. Обнаруживают характерную дилатацию ПЖ в сочетании с региональными нарушениями сократительной способности или дискинезией по типу выпячиваний и аневризм в области выходного тракта ПЖ, верхушки или трикуспидального клапана. Эти же признаки, имеющие специфичность 90%, можно обнаружить и с помощью таких неинвазивных методов исследования, как МРТ сердца и ЭхоКГ. МРТ позволяет точно охарактеризовать функцию и анатомию ПЖ, но этот метод имеет ограниченные диагностические возможности. Например, невозможно оценить точный размер утолщения свободной стенки ПЖ и количество жировой ткани по сравнению с ее обычным содержанием в эпикарде и перикарде (рис. 18.25). В настоящее время ЭхоКГ - наиболее ценный метод, поскольку позволяет провести дифференциальную диагностику, исключить другие заболевания сердца, уточнить функциональную способность ПЖ и ЛЖ и обнаружить их структурные аномалии. ЭхоКГ широко применяют для ранней диагностики прогрессирования заболевания, а также для скринингового обследования членов семьи пациента. При малых формах АКМП локализацию структурных изменений ПЖ обнаружить достаточно сложно. К сожалению, незначительные отклонения от нормы можно не диагностировать даже при использовании всех методов исследования.
Обнаружение типичных гистологических изменений in vivo при АКМП с помощью эндомиокардиальной биопсии имеет определенную диагностическую ценность, но ограничено низкой чувствительностью метода (особенно при невыраженных формах АКМП). Эндомиокардиальная биопсия не может подтвердить трансмуральное замещение миокарда жировой или фиброзно-жировой тканью. Более того, фокальные или сегментарные изменения, отсутствие вовлечения в патологический процесс МЖП и естественное присутствие жировой ткани у пожилых пациентов может привести к гипердиагностике заболевания на основании результатов
биопсии. Повышенное содержание жировой или фиброзной ткани у пожилых пациентов или другие признаки КМП также снижают диагностическую ценность МРТ.
На основании клинических и морфологических изменений для АКМП были установлены диагностические критерии, представленные в табл. 18.10 [217]. Диагноз АКМП базируется на обнаружении двух больших критериев, или одного большого и двух малых критериев, или четырех малых критериев, подтвержденных гистологическими, ЭКГ-находками и существованием аритмии [218].
Таблица 18.10. Большие и малые критерии диагностики аритмогенной кардиомиопатии*
Большие критерии
Семейное заболевание, подтвержденное при аутопсии или хирургическом вмешательстве
Существование волны эпсилон или удлинение комплекса QRSgt;110 мс в отведениях V1-V3
Выраженная дилатация ПЖ и систолическая дисфункция с или без незначительного вовлечения в патологический процесс ЛЖ
Локальные аневризматические расширения ПЖ (зоны акинезии или дискинезии с диастолическим выбуханием)
Выраженная сегментарная дилатация ПЖ
Замещение миокарда фиброзно-жировой тканью (по результатам эндомиокардиальной биопсии)
Малые критерии
Указания на ВСС в семейном анамнезе в возрасте до 35 лет
Семейный анамнез заболевания, подтвержденного диагностическими критериями
Поздние потенциалы на сигнал-усредненной ЭКГ
Инверсия зубца Т в отведениях V2 и V3 без БПНПГ у пациентов в возрасте старше 12 лет
Тахикардия с формой комплекса, соответствующей БЛНПГ (устойчивали или неустойчивая) на ЭКГ, при ХМ или во время пробы с физической нагрузкой
Более 1 тыс. ЖЭС в сутки при ХМ
Небольшая дилатация ПЖ с или без его дисфункции на фоне сохраненной функции ЛЖ
Незначительная сегментарная дилатация ПЖ
Участки региональной гипокинезии правых отделов
Изменено (с разрешения): McKenna W.J., Thiene G., Nava A. et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy // Br. Heart J. - 1994. - Vol. 71. - P. 215-218.
СТРАТИФИКАЦИЯ РИСКА
Основная цель лечения АКМП - профилактика возникновения жизнеугрожающих нарушений ритма и ВСС. Вероятность развития стресс-индуцированной ФЖ или гемодинамически значимой ЖТ
прогнозировать сложно, особенно у асимптомных молодых людей и атлетов. В настоящее время отсутствуют рекомендации о назначении профилактического лечения пациентам с установленным диагнозом АКМП, минимальными клиническими симптомами или морфологическими изменениями. Асимптомные пациенты с высоким риском возникновения ВСС имеют отягощенный семейный анамнез, желудочковые нарушения ритма в детском или подростковом возрасте или во время занятий спортом, обмороки или вовлечение в патологический процесс ЛЖ. Прогностическая ценность ЭКГ, ХМ, нагрузочных тестов и сигнал-усредненной ЭКГ в качестве предиктора ВСС невысока. Пациентов с клиническими симптомами необходимо обследовать более тщательно и при необходимости проводить ангиографию ЛЖ, программируемую желудочковую стимуляцию и эндомиокардиальную биопсию.
Источник: Кэмм А. Джон, Люшер Томас Ф., Серруис П.В., «Болезни сердца и сосудов.Часть 4 (Главы 16-19)» 2011
А так же в разделе « АРИТМОГЕННАЯ КАРДИОМИОПАТИЯ »
- Болезнь накопления гликогена II типа
- Болезнь накопления гликогена III типа
- Болезнь накопления гликогена IV типа
- Болезнь Фабри
- Поражение сердечно-сосудистой системы
- ЛУЧЕВАЯ БОЛЕЗНЬ
- ЛЕЧЕНИЕ СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТИ
- ЛЕЧЕНИЕ НАРУШЕНИЙ РИТМА
- НЕКЛАССИФИЦИРУЕМАЯ КАРДИОМИОПАТИЯ: НЕКОМПАКТНЫЙ МИОКАРД ЛЕВОГО ЖЕЛУДОЧКА