
Тип наследования является важнейшей и постоянной характеристикой любого моногенного заболева-
Аутосомно-доминантный тип наследования болезни имеет место в тех случаях, когда патологический I он является доминирующим и закономерно приводит к развитию симптоматики даже в гетерозиготном состоянии, т.е. на одной из двух гомологичных неполовых хромосом (аутосом). Обычно при аутосомно-доминантных заболеваниях в браке больного и здорового членов семьи распределение аллелей имеет вид Аа х аа (где А - доминантный мутантный ген, а-рецессивный нормальный ген). Данный тип наследования характеризуется следующими признаками:
Типичный пример родословной, в которой имеет место наследование аутосомно-доминантного заболевания, представлен на рис. 8. Среди неврологических заболеваний аутосомно-доминантный тип наследования характерен для хореи Гентингтона, нейрофиброматоза, эссенциального тремора, торсионной дистонии, ряда форм спиноцеребеллярных атаксий и др.
Доминантные гены обладают различной пенет- рантностъю. Под пенетрантностью понимают вероятность проявления мутантного гена у его носителей. В том случае, когда заболевают все члены семьи, имеющие мутантный ген, пенетрантность гена составляет 100%. При неполной пенетрантности мутантного гена отдельные члены семьи, заведомо являющиеся носителями мутации («облигатные» носители), могут на протяжении всей жизни оставаться клинически здоровыми, но при этом они передают мутантный ген и заболевание своим детям. В таких случаях говорят о «пропуске поколения» в родословной (см. рис. 8, индивидуум Ш-5).
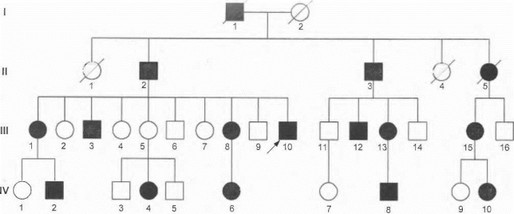
Рис. 8. Родословная семьи с аутосомно-доминантным заболеванием
В целом при отсутствии признаков аутосомно-доминан- тного заболевания у родителей больного возможны следующие объяснения: а) неполная пенетрантность мутан тного гена у одного из родителей, являющегося носителем мутации; б) ложное отцовство; в) возникновение мутации de novo в i амете одного из родителей или у больного на ранней постзиготической стадии.
Еще одним свойством доминантных генов, которое следует принимать во внимание при генеалогическом анализе, является различная экспрессивность. Экспрессивность гена - это степень тяжести и широта спектра клинических проявлений болезни у носителей мутации. Высокая экспрессивность мутантного гена приводит к появлению развернутой клинической картины болезни у лиц, унаследовавших мутантный ген. При низкой экспрессивности гена заболевание может у отдельных носителей мутации проявляться в виде различных «стертых» форм. Так, например, гены аутосомно-доми- нантной торсионной дистонии характеризуются неполной экспрессивностью, при этом у носителей патологического гена могут наблюдаться как' тяжелые генерализованные формы заболевания, так и разнообразные «стертые» варианты (forme fruste), в частности — фокальные дистонические синдромы (кривошея, писчий спазм, блефароспазм), изолированный постуральный тремор рук и т.д. Правильная диагностика таких «стертых» форм болезни в родословной исключительно важна для медико-генетического консультирования, поскольку лица как с развернутой клинической картиной, так и имеющие forme fruste в одинаковой степени являются носителями мутантного гена и могут передать его в следующее поколение, что приведет к появлению новых случаев болезни у потомков.
Аутосомно-рецессивный тип наследования наблюдается при заболеваниях, для манифестации кото-
рых необходимо присутствие мутантного гена в гомозиготном состоянии, т.е. на обеих гомологичных хромосомах, унаследованных от родителей. Чаще всего при ауто- сомно-рецессивных заболеваниях первичный молекулярный дефект заключается в повреждении фермента, а патологический эффект проявляется при кри тическом снижении его активности ниже определенного порогового значения (5-10%) - как это и имеет место при повреждении обеих копий гена. Гетерозиготные носители имеют «одинарную дозу» мутации; благодаря второй (нормальной) копии гена активность белка у них составляет около 50%, что обычно вполне достаточно для поддержания соответствующей функции на физиологическом уровне, поэтому они остаются клинически здоровыми. 11 классических случаях аутосомно-рецессивного наследования генотип родителей больных имеет вид Аа х Аа (где а - рецессивный мутантный ген А - доминантный нормальный ген). Данный тип наследования характеризуется следующими особенностями:
Типичный пример родословной с аутосомно-ре- цессивным заболеванием представлен на рис. 9. Такой тип наследования характерен для болезни Фридрейха, гепатолентикулярной дегенерации, спинальной амиот- рофии Верднига-Гофманаи Кугельберга-Веландер, атаксии-телеангиэктазии и ряда других моногенных заболеваний нервной системы.
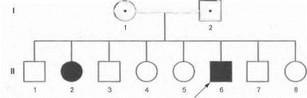
Рис. 9. Родословная семьи с аутосомно-рецессивным заболеванием
При проведении генеалогического анализа в семьях с предполагаемым аутосомно-рецессивным типом наследования следует принимать во внимание одно важное обстоятельство. Как было указано выше, в соответствии с менделевскими законами доля сибсов, пораженных аутосомно-рецессивным заболеванием, должна составлять около % от общего числа детей в поколении. Поскольку для современной структуры семей характерно сравнительно небольшое число детей (1-3 ребенка), в большинстве случаев аутосомно-рецессивные болезни проявляются в виде единичных (снорадических) случаев, и наследственно-семейный характер заболевания бывает очевидным далеко не всегда. В такой ситуации отсутствие семейного анамнеза не снимает вопрос о генетической природе болезни и не исключает 25%-й риск появления ее повторных случаев при рождении других к-гей у данной родительской пары.
Еще один источник ошибок в оценке данного типа наследования проиллюстрирован на рис. 10 на примере оольшой семьи с аутосомно-рецессивной мышечной дистрофией, обследованной нами в одном из горных пзолятов Северного Кавказа. В данной высокоинбред- п ой семье заболевание наблюдается у 12 родственников пв 3 разных поколений, что, на первый взгляд, противоречит модели аутосомно-рецессивного наследования. ()днако ни в одном случае в данной родословной нет 11 пямой передачи болезни от родителей детям, а в пределах каждой конкретной родительской пары характер сегрегации подчиняется всем закономерностям, характерным для аутосомно-рецессивной патологии. Таким образом, сам по себе факт наличия болезни в нескольких поколениях обширной родословной не исключает ауто- сомно-рецессивный тип наследования, и ключевым признаком здесь является манифестация симптомов у части потомства (-25%) при клинически здоровых родителях, я шлющихся облигатными гетерозиготными носителями мутации.
Правило об отсутствии прямой передачи аутосом- по-рецессивной болезни в следующее поколение имеет редкие исключения: такое бывает возможным в ситуации, когда больной вступает в брак либо с другим больным с тем же заболеванием (тип брака аа х аа), либо с гетерозиготным носителем мутации в том же гене (аа х аА). В первом случае все дети унаследуют 2 копии мутантного гена и будут больными, во втором случае заболеет половина детей.
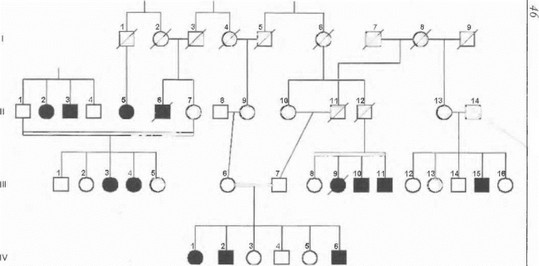
Рис. 10. Родословная большой семьи с аутосомно-рецессивным заболеванием, манифестирующим у ряда родственников из 3 поколений
Глава 1
Общие принципы Генодиагностики
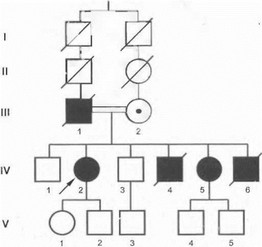
Пример представлен на рис. 11 (показана родословная наблюдавшейся нами семьи, отягощенной болезнью Фридрейха).
В данной семье больной отец (III-1) вступил в кровнородственный брак с троюродной сестрой, являющейся гетерозиготным носителем мутантной хромосомы, унаследованной от общего предка (носительство мутации подтверждено при ДНК-исследовании). В результате заболевание манифестировало у отца и 3 его детей, т.е. в 2 последовательных поколениях. Такой особый характер передачи аутосомно-рецессивного заболевания называется псевододоминантным. В отличие от истинного аутосомно-доминантного типа передачи бо
лезни при псевдодоминантном наследовании болезнь регистрируется обычно лишь в 2 поколениях и не затрагивает серии последовательных поколений и боковых ветвей родословной. Еще одним признаком псевдодоми- нантного наследования является то, что оно чаще всего имеет место в случаях кровнородственных браков, поскольку в соответствующих семьях частота носительства мутантного рецессивного гена среди родственников значительно выше общепопуляционной. Накопец, при псевдодоминантном наследовании число пораженных сиб- сов в каждом поколении больше обычной для аутосом- но-рецессивного наследования цифры 25%.
При локализации мутантного гена в Х-хромосо- ме имеет место наследование, сцепленное с полом. В абсолютном большинстве случаев такие гены являются рецессивными, а тип наследования заболевания - Х-сцеп- ленным рецессивным. Поскольку X и Y-хромосомы не комплементарны, у мужчин даже рецессивный ген, расположенный на единственной Х-хромосоме, не имеет своей пары (состояние гемизиготности) и является манифестирующим; напротив, у женщин-гетерозит мутация на одной из Х-хромосом компенсируется нормальным геном, расположенным на второй копии Х-хромо- сомы. Таким образом, при Х-сцепленном рецессивном типе наследования заболевание проявляется у мужчин, унаследовавших от матери мутантную хромосому. Данный тип наследования имеет следующие признаки:
Пример родословной с Х-сцепленным рецессивным заболеванием показан на рис. 12.
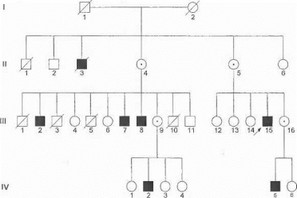
Рис. 12. Родословная семьи с Х-сцепленным рецессивным заболеванием
Х-сцепленное рецессивное наследование свойственно прогрессирующей мышечной дистрофии Дю- I пенна и Бекера, адренолейкодистрофии, спинально-бульбарной амиотрофии Кеннеди и некоторым другим наследственным неврологическим заболеваниям. В связи с тем, что женщины-носительницы мутантного гена обычно остаются клинически здоровыми (пропуск поколений в родословной) и передают болезнь лишь половине сыновей, при Х-сцепленном типе наследования проследить семейный характер болезни бывает весьма непросто, а число спорадических случаев намного превышает частоту семейных форм. Об этом следует помнить при обследовании больных и проведении медико-генетического консультирования.
Иногда у женщин носительство мутантного гена на одной из Х-хромосом не компенсируется присутствием нормально функционирующей копии гена на второй Х-хромосоме. Это случается, например, при различных цитогенетических нарушениях - синдроме Шерешевс- кого -Тернера (генотип ХО, т.е. отсутствие одной Х-хро- мосомы), транслокации критического участка Х-хромо- сомы, а также при высокой частоте инактивации нормальной Х-хромосомы (феномен аномальной лайониза- ции). Такие женщины являются манифестными носи- телъцами гена; у них может наблюдаться развернутая клиническая картина или отдельные симптомы соответствующего Х-сцепленного заболевания, что приводит к серьезным затруднениям в клинической диагностике. Случаи манифестного носительства у женщин являются исключительно редкими (не более 1-2% от общего числа больных с Х-сцепленными рецессивными заболеваниями) и нуждаются в соответствующем подтверждении с использованием цитогенетических методов и методов ДНК-анализа.
В тех случаях, когда ген, локализованный на X- хромосоме, определяет развитие доминантного признака, имеет место Х-сцепленный доминантный тип наследования. Для него характерны следующие особенности:
Пример родословной с Х-сцепленным доминан- шым типом наследования представлен на рис. 13.
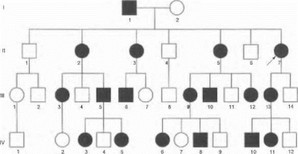
Рис. 13. Родословная семьи с Х-сцепленным доминантным заболеванием
Данный тип наследования является исключительно редким. В неврологии практически единственным примером заболевания с Х-сцепленным доминантным типом наследования является наследственная моторносенсорная демиелинизирующая невропатия, обусловленная мутациями в гене Сх32.
Все рассмотренные выше классические менделев- ские типы наследования свойственны заболеваниям, обусловленным мутациями ядерной ДНК; их закономер- кости определяются механизмом мейотического расхождения хромосом половых клеток в процессе деления клеточного ядра. В 80-90-е годы был открыт новый класс наследственных болезней человека - митохондриальные цитопатии. Они вызываются мутациями генов в составе коротких кольцевых молекул ДНК, расположенных в митохондриях- т.е. в органеллах цитоплазмы, вне клеточного ядра (раздел 1.1). Митохондриальные цитопатии характеризуются совершенно особым, неменделев- ским характером передачи заболевания в родословной, который определяется как митохондриальный (цитоплазматический, неядерный) тип наследования. Поскольку набор митохондрий в клетках организма имеет исключительно материнское происхождение, митохондриальный тип наследования болезни имеет следующие основные признаки:
На рис. 14 представлена типичная родословная семьи с митохондриальным (материнским) наследованием болезни. По такому типу передаются синдромы MERRF (миоклонус-эпилепсия с рваными красными волокнами), MELAS (митохондриальная энцефаломио- патия с лактат-ацидозом и инсультоподобными эпизодами) и некоторые другие заболевания нервной системы. Более подробно клинико-генеалогические особенности группы митохондриальных болезней разбираются в разделе 3.8.
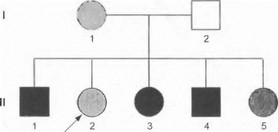
Рис. 14. Родословная семьи с заболеванием, наследующимся по материнскому (митохондриальному) типу Разной штриховкой обозначены больные с различной степенью тяжести заболевания.
Важнейшим этапом обследования больного с подозрением на наследственное заболевание является сбор генеалогического анамнеза. Его целью является составление родословной, позволяющей проследить передачу болезни в ряду поколений, установить тип наследования болезни и определить круг лиц, принадлежащих к группе риска и нуждающихся в медико-генетическом консультировании (в том числе с использованием методов ДНК-диагностики).
Можно выделить несколько основных этапов, на которые подразделяется процедура генеалогического анализа в обследуемой семье.
Для получения генеалогической информации может использоваться анкетирование, при этом решающим фактором успеха является адекватный перечень вопросов анкеты и доступность вопросов для членов семьи, не имеющих медицинского образования. Очень важно провести личный осмотр ближайших родственников больного, а в необходимых случаях и других членов семьи с целью более детальной оценки их клинического статуса. При необходимости данные личного осмотра родственников больного могут дополняться
Накопление случаев одного и того же заболева-
Составление родословной проводится от пробанда к ближайшим и отдаленным родственникам (пробанд- лицо, через которое осуществляется регистрация всей семьи). Лица, принадлежащие к одному поколению, располагаются в родословной строго в одном ряду. Каждый член семьи кодируется индивидуальным индексом (1-3, П-4, III-12 и т.д.), отражающим номер поколения (римская цифра, нумерация сверху вниз) и порядковый номер в соответствующем поколении (арабская цифра, слева направо). В некоторых случаях один из представителей той или иной родительской пары может не отображаться в родословной, если он клинически здоров и не имеет прямого отношения к передаче мутантного гена.

1*ис. 15. Основные символы, рекомендуемые к использованию при составлении родословной: 1 - лица мужского пола; 2 - ница женского пола; 3 - пол неизвестен (а-здоровые, б - больные); •I - несколько человек (а - число известно, б - число неизвестно); 5 - пробанд; 6 - умершие; 7 - брак; 8 - родственный брак; 9 - сибсы (дети одной родительской пары); 10 - монозиготные близнецы; 11 - дизиготные близнецы; 12 - облигатный гетерозиготный носитель мутантного гена (клинически здоров и не заболеет); 13 - носитель мутантного гена на пресимптоматической стадии (имеет высокий риск заболеть по достижении соответствующего возраста); 14 - бездетный брак; 15 - выкидыш; 16 - медицинский аборт; 17 - усыновление.
Составленная родословная должна дополняться так называемой легендой - письменным изложением дополнительных сведений, имеющих значение для обследования изучаемой семьи и генеалогического анализа (национальность, место рождения и адреса включенных в родословнзто членов семьи, имеющиеся результаты клинико-инструментального обследования, другая касающаяся родственников информация, полученная из различных медицинских документов, и т.п.).
г) ранняя смерть одного из родителей (который мог не дожить до возраста манифестации болезни и поэтому характеризовался родственниками как «здоровый»);
д) место рождения родителей и более отдаленных предков (происхождение родителей больного и/или их предков из одного и того же сравнительно изолированного населенного пункта может косвенно свидетельствовать о возможности определенной кровнородственной связи между ними) и т.д. При наличии нескольких случаев заболевания в семье и несоответствии типа передачи какой-либо определенной модели наследования (менделев- ской или митохондриальной) может быть сделан вывод
Следует подчеркнуть, что правильно установлен- ш.гй тип наследования уже сам по себе имеет важное диагностическое значение. В качестве примера можно 111 юдставить типичную ситуацию, при которой в консуль-
- ия; он отражает функциональную значимость соответствующего мутантного гена, его хромосомную локализацию и механизмы реализации мутации на клеточном уровне. Менделирующие заболевания нервной системы могут наследоваться по аутосомно-доминантному, ауто- сомно-рецессивному и Х-сцепленному типам.
Аутосомно-доминантный тип наследования болезни имеет место в тех случаях, когда патологический I он является доминирующим и закономерно приводит к развитию симптоматики даже в гетерозиготном состоянии, т.е. на одной из двух гомологичных неполовых хромосом (аутосом). Обычно при аутосомно-доминантных заболеваниях в браке больного и здорового членов семьи распределение аллелей имеет вид Аа х аа (где А - доминантный мутантный ген, а-рецессивный нормальный ген). Данный тип наследования характеризуется следующими признаками:
- прямая передача болезни от одного из родителей потомкам («вертикальная» передача болезни), в том числе передача заболевания детям от больного отца; нередко может прослеживаться манифестация заболевания в нескольких поколениях;
- соотношение больных и здоровых лиц у потомков больного индивидуума близко к 50%; соответственно, для каждого из детей - потомков больного родителя риск унаследовать мутантный ген (т.е. риск возникновения заболевания) составляет 50%;
- мужчины и женщины поражаются обычно в равной степени; в редких случаях (например, при торсионной дистонии) может наблюдаться более высокая пе- нетрантность гена и более тяжелое течение заболевания у лиц определенного пола - чаще у женщин.
Типичный пример родословной, в которой имеет место наследование аутосомно-доминантного заболевания, представлен на рис. 8. Среди неврологических заболеваний аутосомно-доминантный тип наследования характерен для хореи Гентингтона, нейрофиброматоза, эссенциального тремора, торсионной дистонии, ряда форм спиноцеребеллярных атаксий и др.
Доминантные гены обладают различной пенет- рантностъю. Под пенетрантностью понимают вероятность проявления мутантного гена у его носителей. В том случае, когда заболевают все члены семьи, имеющие мутантный ген, пенетрантность гена составляет 100%. При неполной пенетрантности мутантного гена отдельные члены семьи, заведомо являющиеся носителями мутации («облигатные» носители), могут на протяжении всей жизни оставаться клинически здоровыми, но при этом они передают мутантный ген и заболевание своим детям. В таких случаях говорят о «пропуске поколения» в родословной (см. рис. 8, индивидуум Ш-5).
Рис. 8. Родословная семьи с аутосомно-доминантным заболеванием
В целом при отсутствии признаков аутосомно-доминан- тного заболевания у родителей больного возможны следующие объяснения: а) неполная пенетрантность мутан тного гена у одного из родителей, являющегося носителем мутации; б) ложное отцовство; в) возникновение мутации de novo в i амете одного из родителей или у больного на ранней постзиготической стадии.
Еще одним свойством доминантных генов, которое следует принимать во внимание при генеалогическом анализе, является различная экспрессивность. Экспрессивность гена - это степень тяжести и широта спектра клинических проявлений болезни у носителей мутации. Высокая экспрессивность мутантного гена приводит к появлению развернутой клинической картины болезни у лиц, унаследовавших мутантный ген. При низкой экспрессивности гена заболевание может у отдельных носителей мутации проявляться в виде различных «стертых» форм. Так, например, гены аутосомно-доми- нантной торсионной дистонии характеризуются неполной экспрессивностью, при этом у носителей патологического гена могут наблюдаться как' тяжелые генерализованные формы заболевания, так и разнообразные «стертые» варианты (forme fruste), в частности — фокальные дистонические синдромы (кривошея, писчий спазм, блефароспазм), изолированный постуральный тремор рук и т.д. Правильная диагностика таких «стертых» форм болезни в родословной исключительно важна для медико-генетического консультирования, поскольку лица как с развернутой клинической картиной, так и имеющие forme fruste в одинаковой степени являются носителями мутантного гена и могут передать его в следующее поколение, что приведет к появлению новых случаев болезни у потомков.
Аутосомно-рецессивный тип наследования наблюдается при заболеваниях, для манифестации кото-
рых необходимо присутствие мутантного гена в гомозиготном состоянии, т.е. на обеих гомологичных хромосомах, унаследованных от родителей. Чаще всего при ауто- сомно-рецессивных заболеваниях первичный молекулярный дефект заключается в повреждении фермента, а патологический эффект проявляется при кри тическом снижении его активности ниже определенного порогового значения (5-10%) - как это и имеет место при повреждении обеих копий гена. Гетерозиготные носители имеют «одинарную дозу» мутации; благодаря второй (нормальной) копии гена активность белка у них составляет около 50%, что обычно вполне достаточно для поддержания соответствующей функции на физиологическом уровне, поэтому они остаются клинически здоровыми. 11 классических случаях аутосомно-рецессивного наследования генотип родителей больных имеет вид Аа х Аа (где а - рецессивный мутантный ген А - доминантный нормальный ген). Данный тип наследования характеризуется следующими особенностями:
- болезнь проявляется в одном поколении среди сибсов (т.е. среди братьев-сестер - детей одной родительской пары), родители при этом клинически здоровы («горизонтальная» передача болезни);
- доля пораженных сибсов среди всех потомков определенной родительской пары составляет около 25% (возможные вариации этой цифры в разных семьях обусловлены случайным характером наследования определенной комбинации родительских хромосом); риск развития заболевания у каждого ребенка соответствующей родительской пары также составляет 25%;
- у родителей больных лиц часто имеет место кровнородственный брак (именно в кровнородственном браке наиболее высока вероятность того, что дети унаследуют от обоих родителей две мутантные хромосомы, имеющие общее гепетическое происхождение);
- мужчины и женщины поражаются в равной степени.
Типичный пример родословной с аутосомно-ре- цессивным заболеванием представлен на рис. 9. Такой тип наследования характерен для болезни Фридрейха, гепатолентикулярной дегенерации, спинальной амиот- рофии Верднига-Гофманаи Кугельберга-Веландер, атаксии-телеангиэктазии и ряда других моногенных заболеваний нервной системы.
Рис. 9. Родословная семьи с аутосомно-рецессивным заболеванием
При проведении генеалогического анализа в семьях с предполагаемым аутосомно-рецессивным типом наследования следует принимать во внимание одно важное обстоятельство. Как было указано выше, в соответствии с менделевскими законами доля сибсов, пораженных аутосомно-рецессивным заболеванием, должна составлять около % от общего числа детей в поколении. Поскольку для современной структуры семей характерно сравнительно небольшое число детей (1-3 ребенка), в большинстве случаев аутосомно-рецессивные болезни проявляются в виде единичных (снорадических) случаев, и наследственно-семейный характер заболевания бывает очевидным далеко не всегда. В такой ситуации отсутствие семейного анамнеза не снимает вопрос о генетической природе болезни и не исключает 25%-й риск появления ее повторных случаев при рождении других к-гей у данной родительской пары.
Еще один источник ошибок в оценке данного типа наследования проиллюстрирован на рис. 10 на примере оольшой семьи с аутосомно-рецессивной мышечной дистрофией, обследованной нами в одном из горных пзолятов Северного Кавказа. В данной высокоинбред- п ой семье заболевание наблюдается у 12 родственников пв 3 разных поколений, что, на первый взгляд, противоречит модели аутосомно-рецессивного наследования. ()днако ни в одном случае в данной родословной нет 11 пямой передачи болезни от родителей детям, а в пределах каждой конкретной родительской пары характер сегрегации подчиняется всем закономерностям, характерным для аутосомно-рецессивной патологии. Таким образом, сам по себе факт наличия болезни в нескольких поколениях обширной родословной не исключает ауто- сомно-рецессивный тип наследования, и ключевым признаком здесь является манифестация симптомов у части потомства (-25%) при клинически здоровых родителях, я шлющихся облигатными гетерозиготными носителями мутации.
Правило об отсутствии прямой передачи аутосом- по-рецессивной болезни в следующее поколение имеет редкие исключения: такое бывает возможным в ситуации, когда больной вступает в брак либо с другим больным с тем же заболеванием (тип брака аа х аа), либо с гетерозиготным носителем мутации в том же гене (аа х аА). В первом случае все дети унаследуют 2 копии мутантного гена и будут больными, во втором случае заболеет половина детей.
Рис. 10. Родословная большой семьи с аутосомно-рецессивным заболеванием, манифестирующим у ряда родственников из 3 поколений
Глава 1
Общие принципы Генодиагностики
Пример представлен на рис. 11 (показана родословная наблюдавшейся нами семьи, отягощенной болезнью Фридрейха).
В данной семье больной отец (III-1) вступил в кровнородственный брак с троюродной сестрой, являющейся гетерозиготным носителем мутантной хромосомы, унаследованной от общего предка (носительство мутации подтверждено при ДНК-исследовании). В результате заболевание манифестировало у отца и 3 его детей, т.е. в 2 последовательных поколениях. Такой особый характер передачи аутосомно-рецессивного заболевания называется псевододоминантным. В отличие от истинного аутосомно-доминантного типа передачи бо
лезни при псевдодоминантном наследовании болезнь регистрируется обычно лишь в 2 поколениях и не затрагивает серии последовательных поколений и боковых ветвей родословной. Еще одним признаком псевдодоми- нантного наследования является то, что оно чаще всего имеет место в случаях кровнородственных браков, поскольку в соответствующих семьях частота носительства мутантного рецессивного гена среди родственников значительно выше общепопуляционной. Накопец, при псевдодоминантном наследовании число пораженных сиб- сов в каждом поколении больше обычной для аутосом- но-рецессивного наследования цифры 25%.
При локализации мутантного гена в Х-хромосо- ме имеет место наследование, сцепленное с полом. В абсолютном большинстве случаев такие гены являются рецессивными, а тип наследования заболевания - Х-сцеп- ленным рецессивным. Поскольку X и Y-хромосомы не комплементарны, у мужчин даже рецессивный ген, расположенный на единственной Х-хромосоме, не имеет своей пары (состояние гемизиготности) и является манифестирующим; напротив, у женщин-гетерозит мутация на одной из Х-хромосом компенсируется нормальным геном, расположенным на второй копии Х-хромо- сомы. Таким образом, при Х-сцепленном рецессивном типе наследования заболевание проявляется у мужчин, унаследовавших от матери мутантную хромосому. Данный тип наследования имеет следующие признаки:
- заболевают только мужчины (исключения из этого правила крайне редки и рассматриваются ниже);
- заболевание передается клинически здоровыми женщинами-носителышцами половине сыновей (посредством передачи им мутантной Х-хромосомы);
- отсутствует прямая передача болезни от мужчин их сыновьям (поскольку сыновья всегда наследуют от отца нормальную Y-хромосому);
- все дочери больных-мужчин являются клинически здоровыми гетерозиготными носительницами мутации.
Пример родословной с Х-сцепленным рецессивным заболеванием показан на рис. 12.
Рис. 12. Родословная семьи с Х-сцепленным рецессивным заболеванием
Х-сцепленное рецессивное наследование свойственно прогрессирующей мышечной дистрофии Дю- I пенна и Бекера, адренолейкодистрофии, спинально-бульбарной амиотрофии Кеннеди и некоторым другим наследственным неврологическим заболеваниям. В связи с тем, что женщины-носительницы мутантного гена обычно остаются клинически здоровыми (пропуск поколений в родословной) и передают болезнь лишь половине сыновей, при Х-сцепленном типе наследования проследить семейный характер болезни бывает весьма непросто, а число спорадических случаев намного превышает частоту семейных форм. Об этом следует помнить при обследовании больных и проведении медико-генетического консультирования.
Иногда у женщин носительство мутантного гена на одной из Х-хромосом не компенсируется присутствием нормально функционирующей копии гена на второй Х-хромосоме. Это случается, например, при различных цитогенетических нарушениях - синдроме Шерешевс- кого -Тернера (генотип ХО, т.е. отсутствие одной Х-хро- мосомы), транслокации критического участка Х-хромо- сомы, а также при высокой частоте инактивации нормальной Х-хромосомы (феномен аномальной лайониза- ции). Такие женщины являются манифестными носи- телъцами гена; у них может наблюдаться развернутая клиническая картина или отдельные симптомы соответствующего Х-сцепленного заболевания, что приводит к серьезным затруднениям в клинической диагностике. Случаи манифестного носительства у женщин являются исключительно редкими (не более 1-2% от общего числа больных с Х-сцепленными рецессивными заболеваниями) и нуждаются в соответствующем подтверждении с использованием цитогенетических методов и методов ДНК-анализа.
В тех случаях, когда ген, локализованный на X- хромосоме, определяет развитие доминантного признака, имеет место Х-сцепленный доминантный тип наследования. Для него характерны следующие особенности:
- все дочери больного отца наследуют заболевание; передача заболевания от отца сыну невозможна (сыновья наследуют от отца здоровую Y-хромосому);
- вероятность рождения больного ребенка любого пола от больной матери равна 50%;
- в каждой родословной число больных женщин в 2 раза больше, чем больных мужчин.
Пример родословной с Х-сцепленным доминан- шым типом наследования представлен на рис. 13.
Рис. 13. Родословная семьи с Х-сцепленным доминантным заболеванием
Данный тип наследования является исключительно редким. В неврологии практически единственным примером заболевания с Х-сцепленным доминантным типом наследования является наследственная моторносенсорная демиелинизирующая невропатия, обусловленная мутациями в гене Сх32.
Все рассмотренные выше классические менделев- ские типы наследования свойственны заболеваниям, обусловленным мутациями ядерной ДНК; их закономер- кости определяются механизмом мейотического расхождения хромосом половых клеток в процессе деления клеточного ядра. В 80-90-е годы был открыт новый класс наследственных болезней человека - митохондриальные цитопатии. Они вызываются мутациями генов в составе коротких кольцевых молекул ДНК, расположенных в митохондриях- т.е. в органеллах цитоплазмы, вне клеточного ядра (раздел 1.1). Митохондриальные цитопатии характеризуются совершенно особым, неменделев- ским характером передачи заболевания в родословной, который определяется как митохондриальный (цитоплазматический, неядерный) тип наследования. Поскольку набор митохондрий в клетках организма имеет исключительно материнское происхождение, митохондриальный тип наследования болезни имеет следующие основные признаки:
- заболевание передается от больной матери всем ее детям;
- мужчины и женщины (сыновья и дочери больной матери) поражаются в равной степени;
- передача болезни по мужской линии невозможна.
На рис. 14 представлена типичная родословная семьи с митохондриальным (материнским) наследованием болезни. По такому типу передаются синдромы MERRF (миоклонус-эпилепсия с рваными красными волокнами), MELAS (митохондриальная энцефаломио- патия с лактат-ацидозом и инсультоподобными эпизодами) и некоторые другие заболевания нервной системы. Более подробно клинико-генеалогические особенности группы митохондриальных болезней разбираются в разделе 3.8.
Рис. 14. Родословная семьи с заболеванием, наследующимся по материнскому (митохондриальному) типу Разной штриховкой обозначены больные с различной степенью тяжести заболевания.
Важнейшим этапом обследования больного с подозрением на наследственное заболевание является сбор генеалогического анамнеза. Его целью является составление родословной, позволяющей проследить передачу болезни в ряду поколений, установить тип наследования болезни и определить круг лиц, принадлежащих к группе риска и нуждающихся в медико-генетическом консультировании (в том числе с использованием методов ДНК-диагностики).
Можно выделить несколько основных этапов, на которые подразделяется процедура генеалогического анализа в обследуемой семье.
- Установление наследственной природы болезни. Предположение о наследственном характере того или иного заболевания может быть сделано на основании наличия повторных случаев этого заболевания у родственников обследуемого больного. В процессе опроса больного и его родственников нельзя ограничиваться лишь получением сведений о наличии в семье других случаев «аналогичного заболевания». Следует помнить о том, что для наследственных болезней нервной системы характерен значительный фенотипический полиморфизм, а трактовка членами семьи тех или иных симптомов, имеющих место у родственников, может быть весьма субъективной и ошибочной. Поэтому в целях получения максимально точной информации необходимо интересоваться наличием у родственников любых заболеваний, в особенности сопровождающихся какими-либо неврологическими нарушениями. Это особенно важно для заболеваний, имеющих мультисистемные и мульти- органные проявления. Например, миотоническая дистрофия- сравнительно частое наследственное заболевание с аутосомно-доминантным типом передачи и варьирующей экспрессивностью мутантного гена - в развернутых случаях характеризуется миотоническим феноменом, мышечными атрофиями, кардиомиопатией, катарактой, эндокринными нарушениями и рядом других симптомов; в то же время в некоторых случаях единственным проявлением болезни могут быть катаракта либо нарушение сердечной проводимости. Выявление таких симптомов у родственников больного миотонической дистрофией позволяет заподозрить семейный характер за болевания и предпринять необходимые меры для подтверждения носительства мутации у лиц, имеющих субклинические признаки болезни.
Для получения генеалогической информации может использоваться анкетирование, при этом решающим фактором успеха является адекватный перечень вопросов анкеты и доступность вопросов для членов семьи, не имеющих медицинского образования. Очень важно провести личный осмотр ближайших родственников больного, а в необходимых случаях и других членов семьи с целью более детальной оценки их клинического статуса. При необходимости данные личного осмотра родственников больного могут дополняться
- gt;сзультатами соответствующих лабораторно-инструмен- ыльных методов обследования (ЭЭГ, ЭМГ, рентгеновская и магнитно-резонансная компьютерная томография и др.). При сборе семейного анамнеза следует стремиться использовать и другие источники достоверной медицинской и генеалогической информации, например различную медицинскую документацию (выписки из историй болезни, амбулаторные карты), домовые книги, архивные данные и т.п.
Накопление случаев одного и того же заболева-
- ня в семье может быть не связанным с наследованием какой-либо мутации, а объясняться действием опреде- иенных патогенных факторов, общих для всех членов семьи (особенности пищевого режима, среды обитания и т.д.). С этой точки зрения термины «наследственный» 11 «семейный» применительно к повторным случаям заболевания в семье не всегда являются синонимичными 11 могут иметь различный патогенетический смысл. Следует помнить также о том, что тот или иной клинический синдром (фенотип), напоминающий симптоматику известного наследственного заболевания, может быть следствием ряда других болезней ненаследственной природы. В таком случае говорят о фенокопии наследственного заболевания. Папример, фенокопиями хореи Ген-
- ингтона могут быть ревматическая (малая) хорея, сенильная хорея и другие патологические состояния, что
- динципиально меняет прогноз для больного и членов его семьи и требует проведения тщательного дифференциально-диагностического поиска. Таким образом, детальное выяснение особенностей общего и социального анамнеза, условий жизни, характера трудовой деятельности и других факторов является важнейшим элементом обследования больного с подозрением на наследственное заболевание.
- Составление родословной. Для четкого графического отображения родословной и повышения «информационной емкости» представления генеалогических данных существуют общепринятые правила и унифицированная система условных обозначений. Строгое следование соответствующим рекомендациям при составлении родословных является абсолютно необходимым требованием, позволяющим врачам и другим специалистам, работающим в системе медико-социальной помощи, медицинской статистики, судебной криминалистики и т.д., разговаривать «на одном языке» при анализе различных вопросов наследственной патологии и генетической идентификации. В 1995 году международной рабочей группой по стандартизации родословной были предложены обновленные рекомендации по стандартизованной номенклатуре родословной человека [Bennett R. et al., 1995]. Их следует придерживаться в практической деятельности при представлении генеалогической информации. На рис. 15 приводятся основные символы, используемые в настоящее время при составлении родословных.
Составление родословной проводится от пробанда к ближайшим и отдаленным родственникам (пробанд- лицо, через которое осуществляется регистрация всей семьи). Лица, принадлежащие к одному поколению, располагаются в родословной строго в одном ряду. Каждый член семьи кодируется индивидуальным индексом (1-3, П-4, III-12 и т.д.), отражающим номер поколения (римская цифра, нумерация сверху вниз) и порядковый номер в соответствующем поколении (арабская цифра, слева направо). В некоторых случаях один из представителей той или иной родительской пары может не отображаться в родословной, если он клинически здоров и не имеет прямого отношения к передаче мутантного гена.
1*ис. 15. Основные символы, рекомендуемые к использованию при составлении родословной: 1 - лица мужского пола; 2 - ница женского пола; 3 - пол неизвестен (а-здоровые, б - больные); •I - несколько человек (а - число известно, б - число неизвестно); 5 - пробанд; 6 - умершие; 7 - брак; 8 - родственный брак; 9 - сибсы (дети одной родительской пары); 10 - монозиготные близнецы; 11 - дизиготные близнецы; 12 - облигатный гетерозиготный носитель мутантного гена (клинически здоров и не заболеет); 13 - носитель мутантного гена на пресимптоматической стадии (имеет высокий риск заболеть по достижении соответствующего возраста); 14 - бездетный брак; 15 - выкидыш; 16 - медицинский аборт; 17 - усыновление.
Составленная родословная должна дополняться так называемой легендой - письменным изложением дополнительных сведений, имеющих значение для обследования изучаемой семьи и генеалогического анализа (национальность, место рождения и адреса включенных в родословнзто членов семьи, имеющиеся результаты клинико-инструментального обследования, другая касающаяся родственников информация, полученная из различных медицинских документов, и т.п.).
- Определение типа наследования. После составления родословной и обоснования наследственного характера изучаемого заболевания необходимо установить тип наследования болезни в данной семье. При этом характер сегрегации клинического синдрома сопоставляется с приведенными выше признаками, свойственными всем известным типам наследования. При решении вопроса о типе наследования болезни важно принимать во внимание целый ряд обстоятельств, таких как: а)воз- можность неполной пенетрантности мутантного гена («пропуск поколения»); б) вариабельная экспрессивность мутантного гена (наличие у части родственников «стертых» форм заболевания); в) близкородственные браки;
г) ранняя смерть одного из родителей (который мог не дожить до возраста манифестации болезни и поэтому характеризовался родственниками как «здоровый»);
д) место рождения родителей и более отдаленных предков (происхождение родителей больного и/или их предков из одного и того же сравнительно изолированного населенного пункта может косвенно свидетельствовать о возможности определенной кровнородственной связи между ними) и т.д. При наличии нескольких случаев заболевания в семье и несоответствии типа передачи какой-либо определенной модели наследования (менделев- ской или митохондриальной) может быть сделан вывод
- юм, что данное заболевание является полигенным (мультифакториальным); это означает, что соответствующие члены семьи наследуют не один основной мутан- 111 ый ген, а высокую генетическую предрасположенность, которая может привести либо не привести к бо- незни в зависимости от конкретных модифицирующих факторов среды и индивидуальных особенностей генома.
Следует подчеркнуть, что правильно установлен- ш.гй тип наследования уже сам по себе имеет важное диагностическое значение. В качестве примера можно 111 юдставить типичную ситуацию, при которой в консуль-
- ируемой семье выявлено несколько случаев прогресси-
- gt;у ющей мышечной дистрофии с проксимальным распределением мышечных атрофий и парезов. В случае X- . I ^пленного рецессивного типа наследования такого синдрома следует заподозрить мышечную дистрофию ) (юшенна/Бекера, что требует поиска повреждений в гене или белке дистрофине с помощью ДНК-диагностики и
- ммуногистохимического анализа мышечных биоптатов; с другой стороны, если указанный клинический синд- I х gt;м характеризуется всеми признаками аутосомно-рецес- гивной передачи, врач должен заподозрить диагноз ко- 11ечностно-поясной мышечной дистрофии (формы Эрба- I *ота) и исследовать другие гены и/или белки мышечно- I о волокна. При этом прогноз заболевания и расчеты генетического риска в консультируемой семье будут различными при разных формах патологии.
- Определение круга лиц в изучаемой семье, относящихся к группе риска и нуждающихся в проведении медико-генетического консультирования (т.е. лиц, которые могут являться носителями мутантного гена); расчет конкретной величины генетического риска у консультируемых родственников; осуществление комплекса мер, направленных на профилактику новых случаев заболевания в семье, отягощенной наследственным заболеванием. Детальный анализ этих вопросов в свете современных возможностей ДНК-диагностики представлен в главе 5.