
С момента первого описания и выделения основных групп моногенных наследственных заболеваний нервной системы проблема их систематизации и номенклатуры оставалась одной из наиболее сложных и запутанных в клинической неврологии и нейрогенетике. Это обусловлено рядом взаимосвязанных факторов.
Во-первых, для наследственных заболеваний нервной системы характерен исключительно выраженный меж- и внутрисемейный фенотипический полиморфизм [Давиденков С.Н., 1934; Калмыкова Л.Г., 1976; Иванова-Смоленская И.А. и др., 1998 (a); Harding А., 1984; 1993; Davis М. et al., 1993]. Он проявляется развитием самых разнообразных, нередко весьма отличающихся друг от друга неврологических синдромов в рамках одной нозологической формы, в том числе даже у разных членов одной семьи. Вариабельная экспрессивность мутантных генов может приводить к манифестации как развернутых клинических форм болезни, так и развитию атипичных ее вариантов, а также «стертых» или
«фрустрированных» форм (forme fruste). Для многих моногенных неврологических заболеваний (таких, например, как миотоническая дистрофия, ряд форм наследственных атаксий, торсионной дистонии и др.) полиморфизм клинических проявлений и развитие разнообразных атипичных и «стертых» форм является скорее правилом, чем исключением, что значительно осложняет систематизацию изучаемых клинических синдромов.
Во-вторых, морфологической картине наследственных болезней нервной системы также свойствен значительный полиморфизм, отражающий в первую очередь вариабельный характер и тяжесть мутаций у конкретных больных. При ряде заболеваний строгая корреляция между клиническими и морфологическими признаками отсутствует. Напрймер, согласно классификации I?. Konigsmark и L. Weiner (1970), несомненной анатомической картине оливопонтоцеребеллярной атрофии соответствует не менее 5 заметно отличающихся клинических синдромов дегенеративных атаксий. Следует отметить, что в неврологии различные генетические формы заболеваний могут иметь сходные клинико-морфологические проявления; с другой стороны, при некоторых наследственных заболеваниях ЦНС (эссенциальный , ремор, торсионная дистония) на секции какие-либо изменения в веществе мозга отсутствуют. Таким образом, морфологические критерии (даже в сочетании с особенностями клинического синдрома) в большинстве случаен являются недостаточным фундаментом для строгой М1ассификации наследственных заболеваний нервной | истемы.
Наконец, до настоящего времени патогенез большинства форм наследственных неврологических заболеваний изучен недостаточно, с связи с чем отсутствуют биохимические и другие рутинные лабораторно-инструментальные маркеры, позволяющие нровести четкие нозологические границы среди многообразия сходных к типических синдромов.
В самом общем виде все наследственные заболевания нервной системы могут быть подразделены на 2 большие грунпы:
На протяжении столетия наиболее распространенной и сохранившей свое значение в нейрогенетике до настоящего времени была классификация наследственных болезней, построенная по топическом) признаку и отражающая уровень поражения и основной клинический синдром заболевания.
В современном виде данная классификация может быть представлена следующим образом (таблица 2).

Данная классификация носит в определенной мере условный характер и не исчерпывает всего многообразия возможных комбинаций синдромов, встречающихся у больных с наследственной неврологической патологией. Так, при болезни Фридрейха, традиционно рассматриваемой в группе наследственных атаксий, в патологический процесс в одинаковой степени вовлекаются как структуры ЦНС (задние и боковые столбы спинного мозга), так и периферическая нервная система (спинномозговые ганглии, чувствительные волокна периферических нервов). Хорея Гентингтона с полным основанием может быть отнесена как к наследственным экстрапирамидным заболеваниям, так и к наследственным деменциям, поскольку для данного заболевания характерна классическая триада симптомов в виде хореического гиперкинеза, прогрессирующей деменции подкоркового типа и нарушений в эмоционально-волевой сфере. При одной из основных форм первично-дегенеративных семейных деменций - лобно-височной деменции - почти в половине случаев отмечается развитие выраженного синдрома паркинсонизма (так называемая аутосомно-доминантная лобно-височная деменция с паркинсонизмом). Можно привести большое количество таких примеров. Тем не менее представленный подход к классификации наследственных болезней нервной системы остается в целом весьма удобным с практической точки зрения и в достаточной степени проверен временем.
Важным дополнением приведенной классификации является подразделение указанных групп заболеваний на основные типы наследования (аутосомно-доми- нантный, аутосомно-рецессивный, Х-сцепленный доминантный и рецессивный, митохондриальный или материнский), каждому из которых нередко соответствуют определенные особенности клинической картины, способствующие уточнению диагноза.
По мере изучения наследственных заболеваний нервной системы накапливалось все больше данных о том, что даже в рамках конкретных клинико-морфологических форм с определенным типом наследования существует более глубокая гетерогенность, результатом которой является наличие большого числа вариантов болезни, отличающихся друг от друга наличием или отсутствием различных дополнительных симптомов. Стало очевидным, в основе того или иного «единого» фенотипа может лежать повреждение многих генов и их белковых продуктов, по-видимому, имеющих отношение к различным звеньям соответствующего биохимического каскада или клеточной функции [Rosenberg R., 1990]. Данное предположение нашло свое блестящее подтверждение в последние годы в связи с успехами молекулярной генетики и открытием большого числа генов основных групп наследственных заболеваний нервной системы. Эти открытия представили убедительное обоснование положения о генетической гетерогенности наследственных болезней нервной системы. Таким образом, генетическая гетерогенность является одним из ключевых понятий современной нейрогенетики, имеющим важнейшее значение в решении целого ряда теоретических и прикладных проблем данной области клинической неврологии.
Идентификация генов, разработка разнообразных методов ДНК-диагностики и установление феномена генетической гетерогенности наследственных болезней нервной системы позволили на качественно новом уровне решить проблему построения четкой и упорядоченной современной номенклатуры этих заболеваний, приведя к внедрению наиболее совершенного и принципиально нового геномного подхода к классификации. Геномная (генетическая) классификаг^ия. предполагает определение прямой взаимосвязи между конкретной нозологической формой и повреждением определенного гена, лежащим в основе болезни [Hardy J., Hardy- Gwinn К., 1998]. Ярким примером может служить группа аутосомно-доминантных атаксий, систематизация которых до недавнего времени была чрезвычайно затруднена. В последнее десятилетие было показано, что вариабельность клинико-анатомической картины аутосомно-доминантных атаксий обусловлена генетической гетерогенностью данной группы заболеваний. На сегодняшний день установлено сз'ществование, как минимум, 16 самостоятельных генов, локализованных на различных хромосомах, мутации которых обусловливают развитие доминантных атактических синдромов. В результате стала возможной предельно четкая и объективная молекулярно-генетическая классификация доминантных атаксий, основанная на прямой (выявление мутации в конкретном гене) или косвенной ДНК-диагностике (установление сцепления с конкретным хромосомным локусом). При этом каждому генетическому варианту заболевания присваивается соответствующий условный индекс, например: спиноцеребеплярная атаксия 1-го типа (СЦА1, ген расположен на хромосоме 6р22-р23), спипоцеребеллярная атаксия 2-го типа (СЦА2,, хромосома 12q24.1), спиноцеребеллярная атаксия 3-го типа (•СЦАЗ или болезнь Мачадо-Джозеф, хромосома 14q32.1) и т.д. По такому же принципу строится в настоящее время классификация наследственных спастических параплегий (картированы на хромосомах 15 локусов данных заболеваний, для 4 из которых идентифицированы мутантные гены), нейрональных цероид-липофусцинозов (6 хромосомных локусов, в том числе 5 идентифицированных генов), семейных форм первичного паркинсонизма (7 локусов, в том числе 3 идентифицированных гена) и других групп наследственных неврологических заболеваний.
В ряде случаев при построении современной классификации генетическая индексация дополняется обозначением типа наследования и некоторых других базовых клинических признаков. Так, в группе аутосомных форм конечностно-поясной прогрессирующей мышечной дистрофии (КПМД) цифровой индекс обозначает тип наследования (1 - аутосомио-доминантный, 2 - аутосом- но-рецессивный), а буквенный - конкретный хромосомный локус. Например, символ Ю ТМД 1В обозначает ауто- сомно-доминантную форму конечностно-поясной мышечной дистрофии, ген которой расположен на хромосоме 1 q 11 —q21, а символ КПМД 2D - аутосомно-ренес- сивную форму, обусловленную мутацией гена а-сарког- ликана на хромосоме 17q 12—q21.33. Аналогичным образом, группу наследственных моторно-сенсорных невропатий принято условно подразделять на демиелини- зирующие (тип I) и аксональные формы (тип И), в рамках каждой из которых выделяются отдельные генетические подтипы, например: НМС.Н 1А (или СМТ 1А - от англ. Charcol-Marie-Footh) - обусловлена мутациями гена РМР22 на хромосоме 17р 11.2; НМСН 1В (СМТ 1В) - мутации гена Р0 па хромосоме 1 q22-23; НМСН 2В (СМТ 2В) - аксональная форма, сцепленная с хромосомой 3q 13—22 и т.д. Эти примеры ясно показывают, что для успеха ДНК -диагностики и практической реализации геномного подхода к классификации наследственных заболеваний нервной системы чрезвычайно важным является тесное сотрудничество клинициета-иевролога и врача лаборатории, осуществляющего мутационный скрининг. Квалифицированный клинико-генеалогический анализ синдрома является краеугольным камнем, на котором основаны постановка показаний к проведению ДНК-диагностики, отбор генов, подлежащих исследованию, последовательность процедур мутационного скрининга, а также адекватная трактовка получаемых результатов (рис. 31).
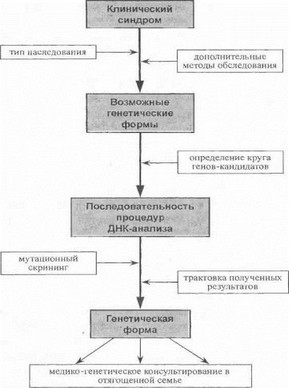
Рис. 31. Общий алгоритм ДНК-диагностики
В наиболее полном виде геномная номенклатура наследственных заболеваний нервной системы представлена в Приложении 1.
Важно подчеркнуть, что вопрос геномной классификации наследственных болезней не носит чисто академический характер, а имеет ярко выраженную практическую направленность. Проведение ДНК-диагности- ки у лиц из группы риска, а также пренатальной ДНК- диагностики с целью профилактики повторных случаев заболевания в семье, может осуществляться только при условии идентификации конкретной молекулярной формы заболевания у пробанда. В будущем при разработке эффективных методов лечения основных групп наследственных заболеваний нервной системы (особенно с использованием методов генной инженерии) точный ДНК- диагноз в каждом конкретном случае будет основным условием для проведения такой специфической терапии. Весьма показательным примером является болезнь Фридрейха - аутосомно-рецессивная форма наследственных атаксий, обусловленная повреждением гена фратак- сина на хромосоме 9ql3. После внедрения в практику прямой ДНК-диагностики было установлено, что целый ряд редких атактических синдромов, заметно отличающихся от «классической» болезни Фридрейха (например, случаи с поздним началом, сохранностью сухожильных рефлексов и т.д.), могут быть обусловлены «мягкими» мутациями в гене фратаксина [Durr A. et al., 1996; Илла- риошкин С.Н. и соавт., 1999 (а)]. Это позволило существенно расширить нозологические границы болезни Фридрейха, пересмотреть существовавшие представления о распространенности данного заболевания, изменить подходы к проведению медико-генетического консультирования в группе семейных и спорадических атаксий.
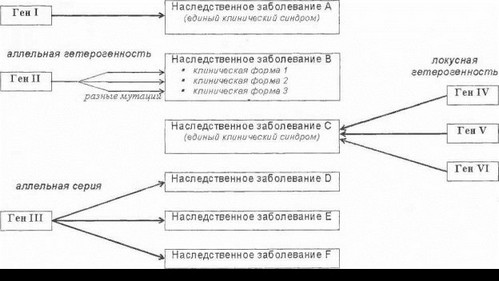
Понятие генетической гетерогенности имеет весьма широкое содержание. Выше были представлены примеры локусной гетерогенности наследственных заболеваний нервной системы. Локусная гетерогенность связана с существованием различных генетических локусов на хромосомах (т.е. различных генов), мутации в которых могут приводить к развитию сходных синдромов. Большое значение имеет также другая разновидность генетической гетерогенности - аллельная гетерогенность, при которой определенное наследственное заболевание может вызываться различными мутациями одного и того же гена (иными словами, у больных могут обнаруживаться различные мутантные аллели одного гена). Приведем несколько неврологических примеров, демонстрирующих ключевую роль аллельной гетерогенности в механизмах фенотипического полиморфизма наследственных болезней нервной системы. Как уже указывалось в главе 1, гепатолентикулярная дегенерация может быть обусловлена большим числом (gt;100) различных точковых мутаций гена медной АТФ-азы, обнаруживаемых практически в любых областях гена. При этом одни мутации в данном гене приводят лишь к снижению функциональной активности фермента различной степени, тогда как другие - к его полной инактивации. В результате тяжесть и характер неврологических и соматических проявлений мутаций у разных больных могут варьировать в весьма широких пределах, что и является одним из факторов выраженного полиморфизма клинической картины гепатолентикулярной дегенерации (5 различных клинических форм болезни, по классификации Н.В. Коновалова). Более ограниченный характер аллельной гетерогенности характерен для заболеваний, обусловленных «динамическими мутациями», поскольку для каждого такого заболевания область мутации в гене строго постоянна (см. главу 1). Так, у всех больных хореей Гентингтона имеет место экспансия три- нуклеотидных повторов CAG в одном и том же участке гена IT-15, однако длина патологически удлиненного тринуклеотидного сегмента может быть различной (от 37 до gt;120 повторов); при этом у больных с тяжелой степенью С AG-экспансии развивается ранний акинети- ко-ригидный вариант Вестфаля, при числе триплетов 45- 55 имеет место классический вариант болезни, а при минимальной степени генетического дефекта (37-40 триплетов) может наблюдаться позднее начало болезни с минимально выраженными нарушениями психики. Таким образом, различные аллели мутантного гена у больных хореей Гентингтона отличаются лишь числом тандемных тринуклеотидных повторов, однако и в данном случае аллельная гетерогенность сопровождается вариабельностью клинических проявлений болезни.
Наличие аллельной гетерогенности нередко приводит к тому, что у больных с аутосомно-рецессивными заболеваниями каждая из двух мутантных хромосом содержит разные мутаций в анализируемом гене. Такое состояние называется компаунд-гетерозиготностыо (от англ, compound - «составной»). Во избежание некоторой терминологической путаницы следует четко подчеркнуть- такие больные являются гомозшотными носителями мутантного гена в том смглслс, что обе копии соответствующего гена повреждены; в то же время они являются компаунд-гетерозиготами (компаундами) применительно к конкретным (разным) мутациям в каждом из двух аллелей гена. «Истинная» гомозиготность при аутосомно рецессивных заболеваниях (т.е. наличие одинаковых мутаций в обоих аллелях гена) обычно имеет место у больных, родившихся от кровнородственного брака и получивших от обоих родителей одну и ту же мутантную хромосому, исторически унаследованную от общего предка.
Еще более сложной является ситуация, когда различные мутации в одном гене могут приводить к развитию совершенно различных клинических фенотипов, квалифицируемых как самостоятельные нозологические формы. Ярким примером такой дивергенции является ген нейрональной а1А-субъединицы потавциал-зависи- мого кальциевого канала, расположенный на хромосоме 19р 13.1. Различные типы мутаций в данном гене ответственны за развитие 3 самостоятельных форм аутосом- но-доминантных наследственных заболеваний нервной системы:
нию мутантным белком новой функции, являются причиной развития семейной гемиплегической мигрени [Carrera Р. et al., 1999;
Tournier-Lasserve Е., 1999].
Хотя указанные клинические формы имеют некоторые общие симптомы (атаксия у больных СЦА6 и ЭА2, пароксизмальность клинических проявлений при ЭА2 и семейной гемиплегической мигрени, наличие атрофии червя мозжечка при всех трех клинических формах), данные фенотипы в клиническом смысле, несомненно, представляют собой самостоятельные заболевания. Эти заболевания являются аллельными, поскольку они обусловлены различными мутантными аллелями единого гена. Такая совокупность аллельных заболеваний, различающихся по своим базовым фенотипическим проявлениям но связанных с повреждением одного гена, называется аллельная серия. Манифестация отличающихся друг от друга заболеваний в рамках каждой аллельной серии объясняется, наиболее вероятно, разными функциональными последствиями соответствующих мутаций (повреждение различных доменов белка, вариабельные механизмы действия мутаций) и рядом других молекулярных факторов (эффект генов-модификаторов, особенности сцепленных гаплотипов и т.д.). Существование аллельных серий является достаточно распространенным феноменом в нейрогенетике, который существенно расширяет наши представления о патогенезе наследственных неврологических заболеваний, функционировании различных биохимических «цепей» нервной системы в норме и при патологии, механизмах формирования конкретных фенотипов. В таблице 3 представлены некоторые наиболее значимые аллельные серии наследственных заболеваний нервной системы.
Таблица 3
Аллельные серии наследственных заболеваний неявной системы
Таким образом, с точки зрения современных знаний, взаимодействие между генотипом и фенотипом носит весьма сложный, комплексный характер, что находит свое преломление в решении различных вопросов диагностики и лечения наследственных заболеваний нервной системы. Указанное взаимодействие в обобщенном виде представлено на рис. 32.
Прогресс в изучении молекулярных основ наследственных заболеваний нервной системы позволяет группировать их по ряду дополнительных признаков - в зависимости от конкретных целей исследования. Так, весьма удобным является подразделение на группы болезней, характеризующихся определенным типом мутаций, например: заболевания с нестабильными «динамическими» мутациями (экспансия гринуклеотидных повторов); заболевания, обусловленные нарушением дозы гена; заболевания с протяженными делециями митохондриальной ДНК и т.д. Такая классификация весьма важна для решения целого ряда теоретических и практических вопросов, поскольку в рамках каждой выделенной группы болезней имеется много общих клинических и патогенетических характеристик (реализация мутаций на клеточном и биохимическом уровне, закономерности фенотипической экспрессии поврежденного гена, методы ДПК-диагностики, подходы к лечению и профилактике).
Более тонкая молекулярно-генетическая субклассификация предполагает подразделение заболевания на фенотипы, соответствующие отдельным му тациям в изучаемом гене. Это имеет основное значение в тех случаях, когда носительство конкретных мутантных аллелей гена достоверно ассоциировано с развитием специфических клинических синдромов (что в нейрогенетике встречается не столь редко). Сущность такой «мутационной» классификации можно проиллюстрировать на примере семейных форм прионных болезней, обуслов-
ленных мутациями гена прионного белка на хромосоме 20р. Как известно, прионные болезни традиционно подразделяются на болезнь Крейтцфельдта-Якоба, синдром Герстманнв-Штреусслера, фатальную семейную инсомнию и куру, однако существование большого числа «промежуточных» клинико-морфологических вариантов существенно осложняет клиническую дифференциацию конкретных синдромов данных заболеваний. На сегодняшний день установлено, что отдельные мутации в гене прионного белка чаще всего приводят к развитию вполне определенных вариантов прионных болезней: например, мутация P102L ассоциирована с синдромом Герст- манна-Штреусслера, мутация Е200К- с особым фенотипом болезни Крейтцфельдта-Якоба, мутация D178N - с фатальной семейной инсомнией [Goldgaber et al., 1989; Collinge J., 1997; Windl O. et al., 1999]. С другой стороны, описан выраженный полиморфизм прионных болезней, обусловленных одной и той же мутацией, что часто не позволяет использовать даже у больных внутри единой родословной какое-то одно традиционное клиническое обозначение [Prusiner S., Hsiao К., 1994]. В связи с этим высказывается предположение о том, что классификация и номенклатура семейных форм прионных болезней должны базироваться главным образом на идеи - тификации конкретной мутации в обследуемой семье, а не на описательных клинико-морфологических терминах [Prusiner S., Hsiao К., 1994; Collinge J., 1997; Windle О. et al., 1999].
Изучение специфической роли бе'лковых продуктов соответствующих мутантных генов знаменует собой появление фунщионалыюго подхода к классификации наследственных заболеваний нервной системы. Данный подход раскрывает патогенетическую сущность болезни и «точку приложения» патологического процесса при конкретных нозологических формах. В зависимости от функции мутантного белка изучаемые заболевания могут быть разделены на 2 большие группы:
В рамках последней группы болезней формируются все новые и новые подгруппы, объединяющие патогенетически сходные нозологические формы, что сопровождается постоянным обновлением и расширением терминологического арсенала нейрогенетики. Так, в самые последние годы было открыто существование особого класса наследственных болезней нервной системы, объединяемых новым термином каналопапши (таблица 4). К ним относятся различные пароксизмальные нервно-мышечные и центральные синдромы (семейные миотонии, периодический паралич, эпизодические атаксии, наследственные формы эпилепсий и др.), обусловленные мутациями генов белковых субъединиц ион-
Молекулярная характеристика группы
каналопатий
Таблица 4
ных каналов - натриевых, калиевых, кальциевых, хлорных [Bulman D., 1997; Jurkat-Rott К. et al., 1999]. Молекулярный анализ каналопатий исключительно важен для установления тонких механизмов проницаемости и возбудимости клеточных мембран, процессов секреции и передачи клеточных сигналов. В качестве других примеров функциональной классификации можно назвать объединение ряда форм прогрессирующих мышечных дистрофий в группу саркогликанопатий (заболеваний, обусловленных мутациями саркогликанов - белков мышечной сарколеммы) [Bushby К., 1999], выделение в рамках нейродегенеративных заболеваний особых групп синуклеинопатий и таупатий (заболеваний, обусловленных патологией а-синуклеин-ассоциированных или гау-ассоциированных белков) [Hardy J., Hardy-Gwinn К., 1998] и т.д. Важно подчеркнуть, что именно функциональный подход к систематизации наследственных неврологических заболеваний служит основой для разработки соответствующих патогенетических методов лечения.
Таким образом, в настоящее время, на рубеже столетий, «остов» наиболее совершенной классификации наследственных заболеваний нервной системы, отражающей молекулярные основы патологического процесса, полностью сформирован. Центральным звеном этой классификации является идентификация первичного молекулярного дефекта (гена, белка и конкретной мутации) у обследуемых больных. При этом генетическая характеристика изучаемого синдрома может дополняться рядом ключевых клинико- генеалогических признаков. Такое построение классификации представляется наиболее оптимальным: во-первых, оно соответствует нуждам повседневной практики, обеспечивая точность диагностики и предусматривая активную роль клиници- став процессе диагностического поиска; во-вторых, оно ориентировано на фундаментальные исследования, имеющие целью разработку патогенетических и этиотропных методов лечения наследственных заболеваний нервной системы.
Во-первых, для наследственных заболеваний нервной системы характерен исключительно выраженный меж- и внутрисемейный фенотипический полиморфизм [Давиденков С.Н., 1934; Калмыкова Л.Г., 1976; Иванова-Смоленская И.А. и др., 1998 (a); Harding А., 1984; 1993; Davis М. et al., 1993]. Он проявляется развитием самых разнообразных, нередко весьма отличающихся друг от друга неврологических синдромов в рамках одной нозологической формы, в том числе даже у разных членов одной семьи. Вариабельная экспрессивность мутантных генов может приводить к манифестации как развернутых клинических форм болезни, так и развитию атипичных ее вариантов, а также «стертых» или
«фрустрированных» форм (forme fruste). Для многих моногенных неврологических заболеваний (таких, например, как миотоническая дистрофия, ряд форм наследственных атаксий, торсионной дистонии и др.) полиморфизм клинических проявлений и развитие разнообразных атипичных и «стертых» форм является скорее правилом, чем исключением, что значительно осложняет систематизацию изучаемых клинических синдромов.
Во-вторых, морфологической картине наследственных болезней нервной системы также свойствен значительный полиморфизм, отражающий в первую очередь вариабельный характер и тяжесть мутаций у конкретных больных. При ряде заболеваний строгая корреляция между клиническими и морфологическими признаками отсутствует. Напрймер, согласно классификации I?. Konigsmark и L. Weiner (1970), несомненной анатомической картине оливопонтоцеребеллярной атрофии соответствует не менее 5 заметно отличающихся клинических синдромов дегенеративных атаксий. Следует отметить, что в неврологии различные генетические формы заболеваний могут иметь сходные клинико-морфологические проявления; с другой стороны, при некоторых наследственных заболеваниях ЦНС (эссенциальный , ремор, торсионная дистония) на секции какие-либо изменения в веществе мозга отсутствуют. Таким образом, морфологические критерии (даже в сочетании с особенностями клинического синдрома) в большинстве случаен являются недостаточным фундаментом для строгой М1ассификации наследственных заболеваний нервной | истемы.
Наконец, до настоящего времени патогенез большинства форм наследственных неврологических заболеваний изучен недостаточно, с связи с чем отсутствуют биохимические и другие рутинные лабораторно-инструментальные маркеры, позволяющие нровести четкие нозологические границы среди многообразия сходных к типических синдромов.
В самом общем виде все наследственные заболевания нервной системы могут быть подразделены на 2 большие грунпы:
- Заболевания, при которых поражение нервной системы составляет ведущий симптомокомп- лекс, «ядро» клинической картины. Именно данные заболевания, которым посвящены пос- ледующие^лавы настоящей монографии, являются основным предметом изучения клинической нейрогенетики.
- Заболевания с системными наследственными дефектами метаболизма, при которых, наряду с поражением различных органов и тканей, в качестве одного из проявлений может наблюдаться вовлечение нервной системы. Классическими примерами являются различные «болезни накопления» (гликогенозы, лизосом- ные болезни), наследственные дефекты обмена аминокислот, пуринов, порфиринов и т.д. В большинстве случаев данные заболевания встречаются в педиатрической практике.
На протяжении столетия наиболее распространенной и сохранившей свое значение в нейрогенетике до настоящего времени была классификация наследственных болезней, построенная по топическом) признаку и отражающая уровень поражения и основной клинический синдром заболевания.
В современном виде данная классификация может быть представлена следующим образом (таблица 2).
Данная классификация носит в определенной мере условный характер и не исчерпывает всего многообразия возможных комбинаций синдромов, встречающихся у больных с наследственной неврологической патологией. Так, при болезни Фридрейха, традиционно рассматриваемой в группе наследственных атаксий, в патологический процесс в одинаковой степени вовлекаются как структуры ЦНС (задние и боковые столбы спинного мозга), так и периферическая нервная система (спинномозговые ганглии, чувствительные волокна периферических нервов). Хорея Гентингтона с полным основанием может быть отнесена как к наследственным экстрапирамидным заболеваниям, так и к наследственным деменциям, поскольку для данного заболевания характерна классическая триада симптомов в виде хореического гиперкинеза, прогрессирующей деменции подкоркового типа и нарушений в эмоционально-волевой сфере. При одной из основных форм первично-дегенеративных семейных деменций - лобно-височной деменции - почти в половине случаев отмечается развитие выраженного синдрома паркинсонизма (так называемая аутосомно-доминантная лобно-височная деменция с паркинсонизмом). Можно привести большое количество таких примеров. Тем не менее представленный подход к классификации наследственных болезней нервной системы остается в целом весьма удобным с практической точки зрения и в достаточной степени проверен временем.
Важным дополнением приведенной классификации является подразделение указанных групп заболеваний на основные типы наследования (аутосомно-доми- нантный, аутосомно-рецессивный, Х-сцепленный доминантный и рецессивный, митохондриальный или материнский), каждому из которых нередко соответствуют определенные особенности клинической картины, способствующие уточнению диагноза.
По мере изучения наследственных заболеваний нервной системы накапливалось все больше данных о том, что даже в рамках конкретных клинико-морфологических форм с определенным типом наследования существует более глубокая гетерогенность, результатом которой является наличие большого числа вариантов болезни, отличающихся друг от друга наличием или отсутствием различных дополнительных симптомов. Стало очевидным, в основе того или иного «единого» фенотипа может лежать повреждение многих генов и их белковых продуктов, по-видимому, имеющих отношение к различным звеньям соответствующего биохимического каскада или клеточной функции [Rosenberg R., 1990]. Данное предположение нашло свое блестящее подтверждение в последние годы в связи с успехами молекулярной генетики и открытием большого числа генов основных групп наследственных заболеваний нервной системы. Эти открытия представили убедительное обоснование положения о генетической гетерогенности наследственных болезней нервной системы. Таким образом, генетическая гетерогенность является одним из ключевых понятий современной нейрогенетики, имеющим важнейшее значение в решении целого ряда теоретических и прикладных проблем данной области клинической неврологии.
Идентификация генов, разработка разнообразных методов ДНК-диагностики и установление феномена генетической гетерогенности наследственных болезней нервной системы позволили на качественно новом уровне решить проблему построения четкой и упорядоченной современной номенклатуры этих заболеваний, приведя к внедрению наиболее совершенного и принципиально нового геномного подхода к классификации. Геномная (генетическая) классификаг^ия. предполагает определение прямой взаимосвязи между конкретной нозологической формой и повреждением определенного гена, лежащим в основе болезни [Hardy J., Hardy- Gwinn К., 1998]. Ярким примером может служить группа аутосомно-доминантных атаксий, систематизация которых до недавнего времени была чрезвычайно затруднена. В последнее десятилетие было показано, что вариабельность клинико-анатомической картины аутосомно-доминантных атаксий обусловлена генетической гетерогенностью данной группы заболеваний. На сегодняшний день установлено сз'ществование, как минимум, 16 самостоятельных генов, локализованных на различных хромосомах, мутации которых обусловливают развитие доминантных атактических синдромов. В результате стала возможной предельно четкая и объективная молекулярно-генетическая классификация доминантных атаксий, основанная на прямой (выявление мутации в конкретном гене) или косвенной ДНК-диагностике (установление сцепления с конкретным хромосомным локусом). При этом каждому генетическому варианту заболевания присваивается соответствующий условный индекс, например: спиноцеребеплярная атаксия 1-го типа (СЦА1, ген расположен на хромосоме 6р22-р23), спипоцеребеллярная атаксия 2-го типа (СЦА2,, хромосома 12q24.1), спиноцеребеллярная атаксия 3-го типа (•СЦАЗ или болезнь Мачадо-Джозеф, хромосома 14q32.1) и т.д. По такому же принципу строится в настоящее время классификация наследственных спастических параплегий (картированы на хромосомах 15 локусов данных заболеваний, для 4 из которых идентифицированы мутантные гены), нейрональных цероид-липофусцинозов (6 хромосомных локусов, в том числе 5 идентифицированных генов), семейных форм первичного паркинсонизма (7 локусов, в том числе 3 идентифицированных гена) и других групп наследственных неврологических заболеваний.
В ряде случаев при построении современной классификации генетическая индексация дополняется обозначением типа наследования и некоторых других базовых клинических признаков. Так, в группе аутосомных форм конечностно-поясной прогрессирующей мышечной дистрофии (КПМД) цифровой индекс обозначает тип наследования (1 - аутосомио-доминантный, 2 - аутосом- но-рецессивный), а буквенный - конкретный хромосомный локус. Например, символ Ю ТМД 1В обозначает ауто- сомно-доминантную форму конечностно-поясной мышечной дистрофии, ген которой расположен на хромосоме 1 q 11 —q21, а символ КПМД 2D - аутосомно-ренес- сивную форму, обусловленную мутацией гена а-сарког- ликана на хромосоме 17q 12—q21.33. Аналогичным образом, группу наследственных моторно-сенсорных невропатий принято условно подразделять на демиелини- зирующие (тип I) и аксональные формы (тип И), в рамках каждой из которых выделяются отдельные генетические подтипы, например: НМС.Н 1А (или СМТ 1А - от англ. Charcol-Marie-Footh) - обусловлена мутациями гена РМР22 на хромосоме 17р 11.2; НМСН 1В (СМТ 1В) - мутации гена Р0 па хромосоме 1 q22-23; НМСН 2В (СМТ 2В) - аксональная форма, сцепленная с хромосомой 3q 13—22 и т.д. Эти примеры ясно показывают, что для успеха ДНК -диагностики и практической реализации геномного подхода к классификации наследственных заболеваний нервной системы чрезвычайно важным является тесное сотрудничество клинициета-иевролога и врача лаборатории, осуществляющего мутационный скрининг. Квалифицированный клинико-генеалогический анализ синдрома является краеугольным камнем, на котором основаны постановка показаний к проведению ДНК-диагностики, отбор генов, подлежащих исследованию, последовательность процедур мутационного скрининга, а также адекватная трактовка получаемых результатов (рис. 31).
Рис. 31. Общий алгоритм ДНК-диагностики
В наиболее полном виде геномная номенклатура наследственных заболеваний нервной системы представлена в Приложении 1.
Важно подчеркнуть, что вопрос геномной классификации наследственных болезней не носит чисто академический характер, а имеет ярко выраженную практическую направленность. Проведение ДНК-диагности- ки у лиц из группы риска, а также пренатальной ДНК- диагностики с целью профилактики повторных случаев заболевания в семье, может осуществляться только при условии идентификации конкретной молекулярной формы заболевания у пробанда. В будущем при разработке эффективных методов лечения основных групп наследственных заболеваний нервной системы (особенно с использованием методов генной инженерии) точный ДНК- диагноз в каждом конкретном случае будет основным условием для проведения такой специфической терапии. Весьма показательным примером является болезнь Фридрейха - аутосомно-рецессивная форма наследственных атаксий, обусловленная повреждением гена фратак- сина на хромосоме 9ql3. После внедрения в практику прямой ДНК-диагностики было установлено, что целый ряд редких атактических синдромов, заметно отличающихся от «классической» болезни Фридрейха (например, случаи с поздним началом, сохранностью сухожильных рефлексов и т.д.), могут быть обусловлены «мягкими» мутациями в гене фратаксина [Durr A. et al., 1996; Илла- риошкин С.Н. и соавт., 1999 (а)]. Это позволило существенно расширить нозологические границы болезни Фридрейха, пересмотреть существовавшие представления о распространенности данного заболевания, изменить подходы к проведению медико-генетического консультирования в группе семейных и спорадических атаксий.
Понятие генетической гетерогенности имеет весьма широкое содержание. Выше были представлены примеры локусной гетерогенности наследственных заболеваний нервной системы. Локусная гетерогенность связана с существованием различных генетических локусов на хромосомах (т.е. различных генов), мутации в которых могут приводить к развитию сходных синдромов. Большое значение имеет также другая разновидность генетической гетерогенности - аллельная гетерогенность, при которой определенное наследственное заболевание может вызываться различными мутациями одного и того же гена (иными словами, у больных могут обнаруживаться различные мутантные аллели одного гена). Приведем несколько неврологических примеров, демонстрирующих ключевую роль аллельной гетерогенности в механизмах фенотипического полиморфизма наследственных болезней нервной системы. Как уже указывалось в главе 1, гепатолентикулярная дегенерация может быть обусловлена большим числом (gt;100) различных точковых мутаций гена медной АТФ-азы, обнаруживаемых практически в любых областях гена. При этом одни мутации в данном гене приводят лишь к снижению функциональной активности фермента различной степени, тогда как другие - к его полной инактивации. В результате тяжесть и характер неврологических и соматических проявлений мутаций у разных больных могут варьировать в весьма широких пределах, что и является одним из факторов выраженного полиморфизма клинической картины гепатолентикулярной дегенерации (5 различных клинических форм болезни, по классификации Н.В. Коновалова). Более ограниченный характер аллельной гетерогенности характерен для заболеваний, обусловленных «динамическими мутациями», поскольку для каждого такого заболевания область мутации в гене строго постоянна (см. главу 1). Так, у всех больных хореей Гентингтона имеет место экспансия три- нуклеотидных повторов CAG в одном и том же участке гена IT-15, однако длина патологически удлиненного тринуклеотидного сегмента может быть различной (от 37 до gt;120 повторов); при этом у больных с тяжелой степенью С AG-экспансии развивается ранний акинети- ко-ригидный вариант Вестфаля, при числе триплетов 45- 55 имеет место классический вариант болезни, а при минимальной степени генетического дефекта (37-40 триплетов) может наблюдаться позднее начало болезни с минимально выраженными нарушениями психики. Таким образом, различные аллели мутантного гена у больных хореей Гентингтона отличаются лишь числом тандемных тринуклеотидных повторов, однако и в данном случае аллельная гетерогенность сопровождается вариабельностью клинических проявлений болезни.
Наличие аллельной гетерогенности нередко приводит к тому, что у больных с аутосомно-рецессивными заболеваниями каждая из двух мутантных хромосом содержит разные мутаций в анализируемом гене. Такое состояние называется компаунд-гетерозиготностыо (от англ, compound - «составной»). Во избежание некоторой терминологической путаницы следует четко подчеркнуть- такие больные являются гомозшотными носителями мутантного гена в том смглслс, что обе копии соответствующего гена повреждены; в то же время они являются компаунд-гетерозиготами (компаундами) применительно к конкретным (разным) мутациям в каждом из двух аллелей гена. «Истинная» гомозиготность при аутосомно рецессивных заболеваниях (т.е. наличие одинаковых мутаций в обоих аллелях гена) обычно имеет место у больных, родившихся от кровнородственного брака и получивших от обоих родителей одну и ту же мутантную хромосому, исторически унаследованную от общего предка.
Еще более сложной является ситуация, когда различные мутации в одном гене могут приводить к развитию совершенно различных клинических фенотипов, квалифицируемых как самостоятельные нозологические формы. Ярким примером такой дивергенции является ген нейрональной а1А-субъединицы потавциал-зависи- мого кальциевого канала, расположенный на хромосоме 19р 13.1. Различные типы мутаций в данном гене ответственны за развитие 3 самостоятельных форм аутосом- но-доминантных наследственных заболеваний нервной системы:
- экспансия тринуклеотидных CAG-повторов в кодирующей области гена приводит к манифестации одной из форм доминантных атаксий - спиноцеребеллярной атаксии 6-го типа (СЦА6); при этом заболевание характеризуется развитием изолированной медленно прогрессирующей атаксии и атрофией верхних отделов полушарий и червя мозжечка [Zhuchenlco О. et al., 1997];
- точковые нонсенс-мутации в данном гене с преждевременным обрывом трансляции являются причиной развития так называемой эпизодической атаксии 2-го типа (ЭА2); она характеризуется пароксизмальными кратковременными приступами атаксии, головокружения, тошноты и диплопии, которые нередко купируются ацетазоламидом [Ophoff R. et al., 1996; Tournier-Lasserve E., 1999].
- наконец, точковые миссенс-мутации в этом же гене, ведущие к замене аминокислоты в составе белка и, предположительно, приобрете
нию мутантным белком новой функции, являются причиной развития семейной гемиплегической мигрени [Carrera Р. et al., 1999;
Tournier-Lasserve Е., 1999].
Хотя указанные клинические формы имеют некоторые общие симптомы (атаксия у больных СЦА6 и ЭА2, пароксизмальность клинических проявлений при ЭА2 и семейной гемиплегической мигрени, наличие атрофии червя мозжечка при всех трех клинических формах), данные фенотипы в клиническом смысле, несомненно, представляют собой самостоятельные заболевания. Эти заболевания являются аллельными, поскольку они обусловлены различными мутантными аллелями единого гена. Такая совокупность аллельных заболеваний, различающихся по своим базовым фенотипическим проявлениям но связанных с повреждением одного гена, называется аллельная серия. Манифестация отличающихся друг от друга заболеваний в рамках каждой аллельной серии объясняется, наиболее вероятно, разными функциональными последствиями соответствующих мутаций (повреждение различных доменов белка, вариабельные механизмы действия мутаций) и рядом других молекулярных факторов (эффект генов-модификаторов, особенности сцепленных гаплотипов и т.д.). Существование аллельных серий является достаточно распространенным феноменом в нейрогенетике, который существенно расширяет наши представления о патогенезе наследственных неврологических заболеваний, функционировании различных биохимических «цепей» нервной системы в норме и при патологии, механизмах формирования конкретных фенотипов. В таблице 3 представлены некоторые наиболее значимые аллельные серии наследственных заболеваний нервной системы.
Таблица 3
Аллельные серии наследственных заболеваний неявной системы
|
Мутантный ген и белок (хромосома) |
Аллельные серин заболеваний |
|
aiA-субъединица потенциалзависимого кальциевого канала (19р13.1) |
|
|
дистрофии (Хр21.2) |
* прогрессирующая мышечная дистрофия Дюшенна;
|
|
РМР22 (17р 11.2) |
|
|
андрогенный рецептор (Xql 1.2—ql2) |
|
|
ГТФ-циклогидролаза 1 (14q22) |
|
|
дисферлин (2 р13) |
|
|
а-субъединица мышечного натриевого канала (17 q 13.1 —cj 13.3) |
|
|
дигидропиридиновый рецептор кальциевого канала (Iq31-q32) |
|
|
протеолипидный белок (Xq22) |
|
|
белок-прекурсор [)- амилоида (21q21) |
|
Таким образом, с точки зрения современных знаний, взаимодействие между генотипом и фенотипом носит весьма сложный, комплексный характер, что находит свое преломление в решении различных вопросов диагностики и лечения наследственных заболеваний нервной системы. Указанное взаимодействие в обобщенном виде представлено на рис. 32.
Прогресс в изучении молекулярных основ наследственных заболеваний нервной системы позволяет группировать их по ряду дополнительных признаков - в зависимости от конкретных целей исследования. Так, весьма удобным является подразделение на группы болезней, характеризующихся определенным типом мутаций, например: заболевания с нестабильными «динамическими» мутациями (экспансия гринуклеотидных повторов); заболевания, обусловленные нарушением дозы гена; заболевания с протяженными делециями митохондриальной ДНК и т.д. Такая классификация весьма важна для решения целого ряда теоретических и практических вопросов, поскольку в рамках каждой выделенной группы болезней имеется много общих клинических и патогенетических характеристик (реализация мутаций на клеточном и биохимическом уровне, закономерности фенотипической экспрессии поврежденного гена, методы ДПК-диагностики, подходы к лечению и профилактике).
Более тонкая молекулярно-генетическая субклассификация предполагает подразделение заболевания на фенотипы, соответствующие отдельным му тациям в изучаемом гене. Это имеет основное значение в тех случаях, когда носительство конкретных мутантных аллелей гена достоверно ассоциировано с развитием специфических клинических синдромов (что в нейрогенетике встречается не столь редко). Сущность такой «мутационной» классификации можно проиллюстрировать на примере семейных форм прионных болезней, обуслов-
ленных мутациями гена прионного белка на хромосоме 20р. Как известно, прионные болезни традиционно подразделяются на болезнь Крейтцфельдта-Якоба, синдром Герстманнв-Штреусслера, фатальную семейную инсомнию и куру, однако существование большого числа «промежуточных» клинико-морфологических вариантов существенно осложняет клиническую дифференциацию конкретных синдромов данных заболеваний. На сегодняшний день установлено, что отдельные мутации в гене прионного белка чаще всего приводят к развитию вполне определенных вариантов прионных болезней: например, мутация P102L ассоциирована с синдромом Герст- манна-Штреусслера, мутация Е200К- с особым фенотипом болезни Крейтцфельдта-Якоба, мутация D178N - с фатальной семейной инсомнией [Goldgaber et al., 1989; Collinge J., 1997; Windl O. et al., 1999]. С другой стороны, описан выраженный полиморфизм прионных болезней, обусловленных одной и той же мутацией, что часто не позволяет использовать даже у больных внутри единой родословной какое-то одно традиционное клиническое обозначение [Prusiner S., Hsiao К., 1994]. В связи с этим высказывается предположение о том, что классификация и номенклатура семейных форм прионных болезней должны базироваться главным образом на идеи - тификации конкретной мутации в обследуемой семье, а не на описательных клинико-морфологических терминах [Prusiner S., Hsiao К., 1994; Collinge J., 1997; Windle О. et al., 1999].
Изучение специфической роли бе'лковых продуктов соответствующих мутантных генов знаменует собой появление фунщионалыюго подхода к классификации наследственных заболеваний нервной системы. Данный подход раскрывает патогенетическую сущность болезни и «точку приложения» патологического процесса при конкретных нозологических формах. В зависимости от функции мутантного белка изучаемые заболевания могут быть разделены на 2 большие группы:
- Энзимопатии и патогенетически близкие к ним заболевания с повреждением транспортных белков. Примерами являются: рецессивная форма дофа-зависи- мой дистонии (ген тирозин-гидроксилазы), атаксия с дефицитом витамина Е (ген транспортера а-токоферола), болезнь Фридрейха (ген фратаксина - трансмембранного переносчика железа) и другие преимущественно ауто- сомно-рецессивные болезни. Общим для данной группы бокьезнсй является полное или частичное (ниже критического порога) нарушение функциональной активности мутантного белка, ведущее к соответствующим биохимическим и фенотипическим проявлениям.
- Заболевания, обусловленные повреждением структурных белков клетки (компонентов клеточных мембран, рецепторов, нейрофиламентов и т.д.). Эти заболевания являются наиболее многочисленными в неврологии и могут наследоваться в соответствии с различными типам менделевской передачи.
В рамках последней группы болезней формируются все новые и новые подгруппы, объединяющие патогенетически сходные нозологические формы, что сопровождается постоянным обновлением и расширением терминологического арсенала нейрогенетики. Так, в самые последние годы было открыто существование особого класса наследственных болезней нервной системы, объединяемых новым термином каналопапши (таблица 4). К ним относятся различные пароксизмальные нервно-мышечные и центральные синдромы (семейные миотонии, периодический паралич, эпизодические атаксии, наследственные формы эпилепсий и др.), обусловленные мутациями генов белковых субъединиц ион-
Молекулярная характеристика группы
каналопатий
Таблица 4
|
Заболевание |
Тип вовлекаемого ионного канала |
Хромосомная локализация гена |
|
|
Мышечные ионные каналы |
|
|
Миотонии Томсена и Бекера |
хлорный капал |
7q35 |
|
Врожденная парамиотопия Эйленбурга |
натриевый канал |
17q23 |
|
Калий-индуцируемая/ а цетазол-чувствительная мйотойия |
натриевый канал |
1 7q23 |
|
Периодический парамиотопический паралич |
натриевый канал |
17q23 |
|
Гиперкалиемическая форма периодического паралича |
натриевый капал |
1 7q23 |
|
Врожденная миастения |
натриевый канал |
2q, 17p |
|
Гипокалиемичеекая форма периодического паралича |
кальциевый капал |
lq32 |
|
Злокачественная гипертермия |
кальциевый канал |
1 q32, 19cjJ_3.1 1Vq13 1 |
|
Болезнь центрального стержня |
кальциевый канал |
|
|
|
Нейрональные ионные каналы |
|
|
Доброкачественные семейные неонатальные судороги |
калиевый капал |
8q24, 20ql3.3 |
|
Эпизодическая атаксия 1 -го типа |
калиевый канал |
12p 13 |
|
Пароксизмальный хореоатетоз со спастичностыо |
калиевый канал (?) |
lp |
|
Генерализованная эпилепсия с (|)ебрильными судорогами плюс |
натриевый канал |
ag СЧ — |
|
Эпизодическая атаксия 2-го типа |
кальциевый капал |
19ql 3.1 |
|
Семейная гемиплегическая мигрень |
кальциевый канал |
19q13.1 |
|
| пиноцеребелЯярная атаксия б-го типа |
кальциевый канал |
19ql3.1 |
|
Нобная эпилепсия с ночными пароксизмами |
кальциевый канал |
20ql3.2-13.3 |
|
11диопатическая 1 сперализованная эпилепсия |
кальциевый канал |
2q22-23 |
|
11 а следственная Iплерэксплексия |
хлорный канал |
5q32 |
ных каналов - натриевых, калиевых, кальциевых, хлорных [Bulman D., 1997; Jurkat-Rott К. et al., 1999]. Молекулярный анализ каналопатий исключительно важен для установления тонких механизмов проницаемости и возбудимости клеточных мембран, процессов секреции и передачи клеточных сигналов. В качестве других примеров функциональной классификации можно назвать объединение ряда форм прогрессирующих мышечных дистрофий в группу саркогликанопатий (заболеваний, обусловленных мутациями саркогликанов - белков мышечной сарколеммы) [Bushby К., 1999], выделение в рамках нейродегенеративных заболеваний особых групп синуклеинопатий и таупатий (заболеваний, обусловленных патологией а-синуклеин-ассоциированных или гау-ассоциированных белков) [Hardy J., Hardy-Gwinn К., 1998] и т.д. Важно подчеркнуть, что именно функциональный подход к систематизации наследственных неврологических заболеваний служит основой для разработки соответствующих патогенетических методов лечения.
Таким образом, в настоящее время, на рубеже столетий, «остов» наиболее совершенной классификации наследственных заболеваний нервной системы, отражающей молекулярные основы патологического процесса, полностью сформирован. Центральным звеном этой классификации является идентификация первичного молекулярного дефекта (гена, белка и конкретной мутации) у обследуемых больных. При этом генетическая характеристика изучаемого синдрома может дополняться рядом ключевых клинико- генеалогических признаков. Такое построение классификации представляется наиболее оптимальным: во-первых, оно соответствует нуждам повседневной практики, обеспечивая точность диагностики и предусматривая активную роль клиници- став процессе диагностического поиска; во-вторых, оно ориентировано на фундаментальные исследования, имеющие целью разработку патогенетических и этиотропных методов лечения наследственных заболеваний нервной системы.