
Геномы различных индивидуумов в рамках одного биологического вида идентичны с точки зрения общего набора генов и их тонкой внутренней структуры. В то же время существует значительное число межиндивидуальных различий, связанных с теми или иными вариациями нуклеотидной последовательности ДНК. Таким образом, любой ген (в более широком смысле - любой генетический локус) может существовать в виде различных вариантов - аллелей. Аллели различаются между собой по составу нуклеотидов в определенных, ограниченных участках ДНК. Количество аллелей одного гена может составлять от 2-3 до нескольких десятков.
Любое изменение структуры ДНК, возникающее спонтанно либо индуцированное путем целенаправленного воздействия физическими или химическими факторами, называется мутацией. Мутации ведут к возникновению новых аллелей соответствующих генов и лежат в основе генетической изменчивости в живой природе. Во многих случаях мутация не имеет каких-либо заметных фенотипических последствий, например при ее локализации в функционально незначимых областях молекулы ДНК. Другим примером «молчащей мутации» является такая замена нуклеотида, при которой новый кодон имеет прежний информационный смысл, т.е. кодирует ту же самую аминокислоту и поэтому не меняет структуру белка. Такие нейтральные мутации рассматриваются как нормальные полиморфизмы, они не элиминируются отбором и могут иметь достаточно высокую частоту в популяции. Изучение полиморфизмов ДНК широко используются в генетической идентификации личности и целом ряде базовых методов молекулярногенетического анализа.
В отличие от нейтральных мутаций (полиморфизмов), патологические мутации приводят к нарушению механизма транскрипции/трансляции гена либо к синтезу аномального белкового продукта. В литературе общий термин «мутация» нередко используется применительно именно к таким случаям нарушений структуры I ена, которые имеют несомненное патогенетическое значение. Аналогичным образом, при дальнейшем изложении материала нами под мутациями будут пониматься любые патогенетически значимые повреждения молекулы ДНК, нриводящие к развитию болезни.
Мутации могут быть генными и хромосомными. Хромосомные мутации затрагивают количественный состав либо структуру хромосом. Такие мутации выявляются при цитогенетическом исследовании в процессе проведения световой микроспокопии специально окрашенных клеточных хромосомных препаратов. Хромосомные мутации обычно являются результатом разовых нарушений деления клеточного ядра в мейозе конкретной половой клетки, поэтому вызванные ими хромосомные болезни чаще всего представлены спорадическими случаями. Генные мутации представляют собой более тонкие изменения нуклеотидного состава генов; заболевания, обусловленные генными мутациями, передаются в родословной в соответствии с рассмотренными выше основными типами наследования. Именно генные мута- ттии и связанные с ними моногенные болезни нервной системы являются основным предметом настоящей монографии.
Генные мутации могут быть классифицированы на основании характера изменений нуклеотидного состава гена, а также на основании механизма нарушения функции соответствующего молекулярного продукта. Основные типы повреждений генов при любых наследственных заболеваниях человека в целом идентичны; однако ряду наследственных заболеваний нервной системы свойственны определенные особенности спектра мутаций в соответствующих генах, знание которых необходимо при проведении Д] 1К-диагностики.
Генные мутации могут быть подразделены на точковые и структурные. Толковые мутации (point mutation) затрагивают один нуклеотид либо 1-2 соседних нуклеотида, тогда как к структурным мутациям относятся более протяженные дефекты гена.
Среди толковых мутаций чаще всего встречаются нуклеотидные замены в кодирующей области гена, т.е. в экзонах. В том случае, если нуклеотидная замена сопровождается изменением аминокислотного шифра соответствующего кодона, такая мутация ведет к изменению состава полипептидной цепи вследствие встраивания в нее «ошибочной» аминокислоты. Мутации данного типа называются миссенс-иутгащшя (missense - т.е. с изменением «смысла» повреждаемого кодона). Тяжесть фенотипических проявлений болезни в этом случае будет определяться функциональной значимостью мутантного участка белка; наиболее тяжелые последствия имеют обычно миссенс-мутации, затрагивающие активные центры белковой молекулы. Иногда нуклеотидная замена приводит к замещению информационно значимого кодона на стоп-кодон; в результате этого на мутантном участке соответствующей молекулы мРНК происходит преждевременный обрыв трансляции и образование «усеченной» (truncated), функционально дефектной молекулы белка. Такие нуклеотидные замены обозначаются термином нонсенс-мутации (nonsense - «бессмысленные», нарушающие информационную роль кодона).
Достаточно распространенным типом мутантных аллелей являются аллели, образующиеся в результате выпадения (делеции) или вставки (инсерции) нуклеотидов в кодирующей области гена. Если число нуклеотидов при делециях и вставках некратно трем (например, в случае точковой мутации с выпадением одного нуклеотида), такая мутация приводит к нарушению нормального отсчета кодирующих триплетов, в результате чего все последующие кодоны в полинуклеотидной цепи приобретают другой аминокислотный смысл (рис. 16).
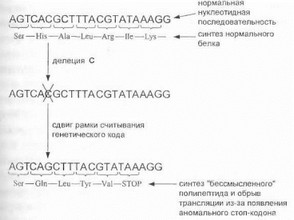
Г’йе. 16. Мутация со сдвигом рамки считывания генстичес- мИ о кода
Другими словами, данный тип мутации сопровождается сдвигом рамки считывания генетического кода (frameshift); мутации со сдвигом рамки ведут к синтезу «бессмысленного» чужеродного белка, который быстро деградирует под воздействием внутриклеточных протеаз.
Если мутация со сдвигом рамки либо нопсенс- мутация расположены в 5’-области гена (т.е. ближе к его началу), они сопровождаются отсутствием синтеза с данной конии гена сколько-нибудь значимой в функциональном отношении полипептидной молекулы и полной потерей активности белка - так называемые нулевые мутации (null mutation). Разумеется, такие мутации являются весьма тяжелыми и нередко либо несовместимы с жизнью, либо характеризуются развитием выраженных мультисистемных проявлений болезни. В неврологии ярким примером такого рода является атаксия-телеангиэктазия, обусловленная повреждением обеих копий гена ATM, кодирующего белок семейства фосфатидили- нозитол-киназ. Около 90% известных на сегодня мутаций в гене ATM представляю-г собой нулевые мутации, приводящие к полной инактивации белка и развитию множественных симптомов поражения головного мозга, кожи, сосудистой, иммунной, эндокринной, костной систем, онкологических осложнений [Gilad S. ei al., 1996].
Толковые и структурные мутации могут происходить не в собственно кодирующих участках гена, а на самом стыке экзонов и интронов - в сайтах сплайсинга (сплайсинговые мутации). Такие мутации нарушают нормальные механизмы созревания первичного РНК- гранс крипта и приводят к неправильному вырезанию нитрона либо к удалению из молекулы РНК информационно значимой экзонной последовательности. В результате этого происходит значительное нарушение структуры белка, ведущее обычно к развитию тяжелых клинических проявлений болезни. В редких случаях мутации мо- гфт затрагивать различные регуляторные последовательности (например, в области промотора или интронов), влияющие на характер экспрессии гена; такие регуляторные мутации обычно сопровождаются не нарушением ыруктуры или функции белковой молекулы, а количественными изменениями содержания белка в клетке.
При некоторых наследственных заболеваниях нервной системы основным типом структурных мутаций являются протяженные делеции, охватывающие шачительную область гена или даже несколько близлежащих генов. Такие делеции, как правило, характеризуются тяжелыми биохимическими и фенотипическими последствиями. Классическими примерами могут служить прогрессир}тощая мышечная дистрофия Дюшен- 11 л/Бекера и аутосомно-рецессивная проксимальная спинальная амиотрофия. Более чем в 60% случаев мышечной дистрофии Дюшенна/Бекера заболевание обусловлено разнообразными делециями гена дистрофина на х | юмосоме Хр21.2, причем тяжесть симптоматики у конкретного больного определяется протяженностью деле- п,1 hi, ее влиянием на рамку считывания кодонов и локализацией по отношению к функционально значимым к (менам дистрофина [Koenig М. е1 al., 1989]. У 98% больных с различными клиническим вариантами аутосомно- рецессивной проксимальной спинальной амиотрофии рюлезни Верднига-Гофмана и Кутельберга-Веландер) обнаруживается деления обеих копий гена SMN на хромосоме 5ql 3, что приводит к отсутствию у больных норма плюго транскрипта данного гена [Lefebvre S. et al., 1995].
При некоторых неврологических заболеваниях I руктурная мутация может заключаться в дупликации
гена или его части. Так, болезнь Пелицеуса-Мерцбахера (одна из форм лейкодистрофий детского возраста) может вызываться дупликацией или толковыми мутациями гена протеолипидного протеина (хромосома Xq22) [Sistermans Е. et al., 1998]. Наиболее частой причиной демиелинизирующей формы наследственной моторносенсорной невропатии (болезнь Шарко -Мари-Тута типа 1 А) является полная дупликация гена РМР22 на хромосоме 17р 11.2 [Lupski J. etal., 1991]. Интересно отметить, что делеция этого же хромосомного участка ведет к развитию совершенно другой клинической формы невропатии - так называемой наследственной невропатии с предрасположенностью к параличам от сдавления [Chance Р. et al., 1993].
В последние годы был открыт принципиально новый тип структурных мутаций, характерный для целого ряда нейродегенеративных заболеваний. Он схематично представлен на рис. 17.

Рис. 17. Экспансия тандемных тринуклеотидных повторов (на примере спиноцеребеллярной атаксии-1)
В рамке показан нуклеотидный состав части гена с областью тринуклеотидных повторов (экзон 1), под рамкой указана соответствующая последовательность аминокислот в составе белка. Тандемным CAG-триплетам гена (выделены жирным шрифтом) соответствует полиглутаминовый участок белка.
Как видно на рисунке, в некоторых участках генов нуклеотидная последовательность представлена цепочкой тандемных тринуклеотидных повторов - например, «цитозии-аденин-гуанин», или (CAG)n. Число таких повторов в норме варьирует в строго определенных пределах; другими словами, существует большое число нормальных аллелей соответствующих генов, отличающихся между собой количеством тандемных тринуклеотидных повторов [Willems R, 1994]. Сущность мутации заключается в патологическом увеличении числа копий данных повторов, превышающем определенные пороговые значения, специфичные для каждой нозологической формы. Данный тип мутаций обозначается как экспансия тринуклеотидных повторов [Иллариошкин С.Н. и др., 1995]. Мутантный удлиненный участок гена является весьма нестабильным, что нередко приводит к дальнейшему изменению (чаще - нарастанию) числа повторов при передаче гена в следующее поколение. В связи с этим мутации по типу экспансии тринуклеотидных повторов получили название динамические мутации [Richards R., Sutherland G., 1992].
Происхождение динамических мутаций представляет собой двухэтапный процесс (рис. 18). На первом этапе в результате редких мутационных событий происходит образование аллелей с «промежуточным» числом тринуклеотидных повторов, превышающим общепопуляционную норму, по не достигающим патологического I трога. Такой а^ГЛель представляют собой «премутацию»: он не приводят к развитию болезни у данного индивидуума, но характеризуется значительной генетической нестабильностью и повышенной вероятностью перехода в «полную мутацию» в последующих поколе- I (мях, что и приводит к манифестации болезни у потом- лgt;в [Richards R., SutherlandG., 1992; Willems R, 1994].
Таким образом, «промежуточные» аллели являются своеобразным резервуаром, постоянным источником новых мутаций в популяции.
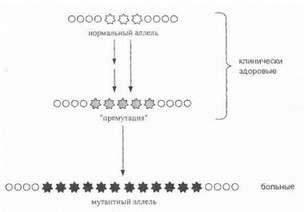
Рис. 18. Двухэтапный механизм происхождения динамических мутаций
Звёздочками обозначены тандемные тринуклеотидные повторы, кружками — другие нуклеотидныепоследовательности гена. Возник ¦ новение аллеля с «промежуточным» числом повторов может быть связано с одним или несколькими мутационными событиями.
К настоящему времени известно уже более десятка наследственных заболеваний нервной системы, обусловленных динамическими мутациями [Rosenberg R., 1996; Brice А., 1998]. Наиболее обширную группу составляют заболевания с аутосомно-доминантным типом наследования (хорея Гентиштона, доминантные атаксии), обусловленные экспансией CAG-повторов в транслируемых областях соответствующих генов. Поскольку триплет CAG кодирует аминокислоту глутамин, такие мутации приводят к пропорциональному удлинению и изменению свойств белковых нолиглутаминовых участков,
вшорме играющих важную роль в реализации сложных межмолекулярных взаймодействий [Housman D., 1995; Ross С., 1995]. Результатом мутации является формирование нерастворимых амилоидогенных белковых комплексов, ведущее к гибели специфических популяций нейронов. Предполагается, что в патогенезе указанной группы болезней важное значение имеет также нарушение взаимодействия мутантных нолиглутаминовых цепей с белками-регуляторами апоптоза, что может приводить к запуску процессов программируемой гибели нейронов | Housman D., 1995; IkedaH. et al., 1996]. Экспансия три- i (уклеотидных повторов может иметь место и в некодирующих областях генов - при атаксии Фридрейха, мио- , онической дистрофии, синдроме ломкой Х-хромосомы. Б этих случаях мутации обусловливают нарушение нормальных механизмов транскрипции гена и угнетение белкового синтеза [Willems R, 1994; Pandolfo М., 1998].
При всех заболеваниях, обусловленных динамическими мутациями, тяжесть клинических проявлений прямо пропорционально коррелирует с величиной экспансии тринуклеотидных повторов, т.е. со степенью тяжести генетического дефекта. Динамический характер мутаций и тенденция к нарастанию числа повторов при передаче гена являются молекулярной основой феномена антиципации, свойственного доминантным «тринук- иеотидным» заболеваниям и заключающегося в появлении все более ранних и тяжелых случаев болезни в каждом последующем поколении [Brice А., 1998]. Было показано также, что необычные свойства динамических мутаций объясняют так называмый эффект отцовской (материнской) передачи, характерный для некоторых из 11 их заболеваний. Так, при хорее Гентингтона наиболее I ижелый акинетико-ригидный вариант болезни (вариант 11естфаля) развивается обычно у детей, унаследовавших оолезнь по отцовской линии; это связано с преимущественной нестабильностью мутантного аллеля и «драматическим» нарастанием числа повторов в мужском хаметогенезе [Telenius Н. et al., 1993; Kremer В. et al., 1995], Напротив, при миотонической дистрофии увеличение степени экспансии повторов наблюдаются обычно при передаче гена по женской линии, в связи с чем случаи тяжелой врожденной миотонической дистрофии наблюдается исключительно при наследовании болезни от матери [Mulley J. et al., 1993]. Данный феномен (зависимость фенотипических проявлений заболевания от пола больного родителя) носит название импринтинг. Различная степень экспансии повторов лежит в основе весьма варьирующей экспрессивности «тринуклеотид- ных» заболеваний - от субклинических и «мягких» форм до тяжелых развернутых случаев с быстрым, фатальпым течением [Иллариошкин С.Н. и др., 1995; 1999].
Некоторые наследственные заболевания нервной системы обусловлены экспансией более сложных по своей структуре повторяющихся участков ДНК, состоящих из десятков нуклеотидов. В их числе можно назвать ми- оклонус-эпилепсию Унферрихта-Лундборга, обусловленную экспансией 12-нуклеотидного повтора в 5’-области гена CST6 на хромосоме 21 q22 [Lafreniere R. et al., 1997; Vitraneva K. et al., 1997], а также семейные формы прионных болезней (болезнь Крейтцфельдта-Якоба), которые в части случаев могут вызываться увеличением копий длинного транслируемого повтора гена прионного белка на хромосоме 20р [Collinge J., 1997].
Для описания различных типов мутаций в том или ином гене разработана специальная номенклатура [BeutlerE., 1993; Beaudet A., Tsui L., 1993; Antonarakis S. et al., 1998]. Она предполагает использование унифицированных символов, позволяющих четко обозначать локализацию мутации в гене, ее характер (толковая мута
ция, делеция, вставка и т.д.), последствия мутации (замена аминокислоты, появление стоп-кодона) и некоторые другие особенности. Номенклатура мутаций носит универсальный характер и позволяет исследователям, работающим в различных областях медицинской генетики, разговаривать «на одном языке» при сопоставлении и анализе даппых мутационного скрининга. Основные элементы действующей номенклатуры генных мутаций человека представлены в Приложении 2.
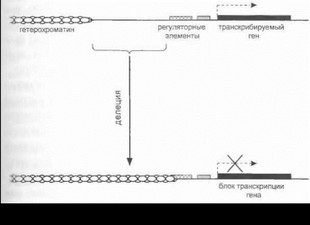
В редких случаях развитие наследственного неврологического заболевания может быть связано не с непосредственными мутациями в гене, а с вторичным нарушением его функции вследствие так называемого негативного позиционного эффекта, или эффекта положения. Примером является лице-лопаточпо-плечевая мышечная дистрофия - аутосомно-доминантпое заболевание, обусловленное делецией вариабельного участка размером 20-250 кб в субтеломерной части хромосомы lq35 [vanDeutekom J. et al., 1993].
Как было показано (рис. 19), мутирующий участок молекулы ДНК не содержит каких-либо генов; однако, делеция этого участка приводит к изменению структуры хроматина в данной хромосомной области и к опосредованному ингибированию транскрипции близлежащего гена (FRG1), расположенного проксимальнее участка разрыва Предполагается, что такой дистантный механизм нарушения экспрессии структурно сохранного гена, связанный с негативным позиционным эффектом крупной внегенной делеции, и обусловливает появление неврологической симптоматики у больных лице-лопаточ- но-плечевой мышечной дистрофией [Fisher J., Upadhyaya М., 1997; Kleinjan D., van Heyningen V., 1998].
По своему патогенетическому механизму все мутации, вызывающие развитие наследственных заболеваний нервной системы, могут быть подразделены на следующие 5 основных групп:
- Мутации, ведущие к потере функции (loss-of- function). К данной группе относятся любые структурные изменения генов (точковые мутации, делеции, экспансия тринуклеотицных повторов в некодирующих областях генов и др.), приводящие к ингибированию процессов транскрипции/трансляции или нарушению нормальной структуры и функциональных свойств белка.
- Мутации, ведущие к появлению новой функции (gain-of-function). Данный тип мутаций, не затрагивая нормальную функцию белка, характеризуется появлением у мутантного протеина новых свойств, обладающих цитотоксичным эффектом и проводящих к постепенной гибели нейронов. Такой механизм характерен для ауто- сомно-доминантных болезней, обусловленных экспансией CAG-повторов в транслируемой области гена: как было рассмотрено выше, удлинение полиглутаминовых участков в составе мутантных белковых молекул сопро- впждается образованием необратимых межмолекулярных «сшивок» и формированием токсичных внутриклеточных полимерных комплексов.
7 Мутации, обладающие доминантным негативным эффектом (dominant negative effect). Доминантный негативный эффект проявляется в том случае, когда продукт мутантного аллеля ингибирует функцию нормальных оелковых молекул. Примером может служить аутосом- I ю-доминантная дофа-зависимая дистония, обусловленная повреждением гена ГТФ-циклогидролазы 1: белковый комплекс данного фермента имеет мультимерную с труктуру и состоит из 10 идентичных субъединиц, по- ттому встраивание в состав комплекса даже одной мушиной молекулы приводит к образованию функционально дефектных гетеромеров и преждевременной деградации фермента [Hirano М. et al., 1998].
I. Мутации, изменяющие «дозу гена» (gene dosage . (feet). Нарgt;шение «дозы гена» имеет место при его де- иениях или дупликациях, что может сопровождаться нарушением нормальной цитоархитектопики соответству- к ниего молекулярного продукта. Так, например, деления пли дупликация гена миелинового белка РМР22 приводит к нарушению внутренней структуры и укладки мие- пша периферических нервов [Chance Р., Tischbeck К., 1994], что сопровождается развитием различных форм наследственных демиелинизирующих невропатий (см. раздел 3.1.3).
з. Мутации, обусловливающие количественные изме
нения первичных молекулярных продуктов гена (мРНК п белка). Такой механизм наблюдается обычно при локализации мутаций в регуляторных областях генов.
Знание механизмов, посредством которых на кле- I очном и биохимическом уровне реализуется патологический эффект различных мутаций, является необходимым условием для разработки новых эффективных методов лечения наследственных заболеваний человека (включая комплексное патогенетическое лечение и разнообразные методы генной терапии).