
Анализ генетических ассоциаций (исследование полиморфизмов в генах-кандидатах) является в настоящее время ведущим подходом при изучении роли генетической составляющей в патогенезе мультифакториальных заболеваний человека, в том числе заболеваний нервной системы [Lander Е., Schork N., 1994]. В его основе лежит тот отмеченный выше факт, что при мультифакториальных болезнях важнейшим фактором наследственной предрасположенности является совокупное действие комплекса генов, определяющих характер ключевых биохимических и иммунологических процессов в организме (особенности метаболизма, характер иммунного ответа, эффективность энергетических реакций и т.д.). Вычленение из общего «генетического фона» наиболее значимых генов (т.е. оказывающих наиболее существенное влияние на вероятность развития того или иного заболевания) и составляет сущность анализа ассоциаций [Lander Е., SchorkN., 1994; Thomson G., 1995 (a); Elston R., 1998; Thomson G., Esposito M., 1999].
Теоретической предпосылкой анализа ассоциаций является существование в большинстве генов человека особых вариабельных участков (полиморфизмов), играющих роль генетических маркеров и различающихся у отдельных индивидуумов по нуклеотидному составу - например, по нейтральной однонуклеотидной замене или по числу коротких нуклеотидных повторов. Таким образом, каждый их этих генов может существовать в виде достаточно большого числа различных вариантов (аллелей). Анализ генетических ассоциаций заключается в сравнении частоты маркерных аллелей определенного гена в группе больных с частотой данных аллелей в общей понуляции. В том случае, если у больных наблюдается достоверное отклонение распределения маркерных аллелей по сравнению с контрольной группой, например достоверное преобладание у них определенного аллеля, делается заключение о наличии ассоциации между данным аллелем гена и изучаемым заболеванием. Таким образом, есть принципиальное различие между рассмотренным в главе 1 генетическим сцеплением и генетической ассоциацией: генетическое сцепление пре^юла- гает взаимосвязь между локусами (маркерным локусом и локусом болезни), тогда как в случае генетической ассоциации речь идет о взаимосвязи заболевания с конкретным аллелем изучаемого генетического локуса.
Возможны 2 основные причины выявленной генетической ассоциации. Во-первых, повышенная (пониженная) частота тех или иных аллелей конкретного гена в группе больных может свидетельствовать о том, что данные варианты гена и его белкового продукта оказывают специфическое модулирующее действие на функционирование биохимических каскадов, определяющих механизм развития патологического процесса. В такой ситуации изучаемый ген имеет прямое этиологическое значение в реализации наследственной предрасположенности к заболеванию, а соответствующие аллели гена могут служить генетическими маркерами и факторами риска (антириска) по данному заболеванию. Во вторых, генетическая ассоциация может быть следствием того, что изучаемый маркер чисто механически находится в тесном сцеплении с одним из «этиологически значимых» генов. В этом случае говорят о неравновесном сцеплении маркерного аллеля и другого, неизвестного гена, ответственного за возникновение заболевания Неравновесное сцепление предполагает, что у значительного большинства больных имеется одна и та же мутантная хромосома, распространившаяся в популяции от общего предка, а изучаемый маркерный аллель не играет самостоятельной патогенетической роли и расположен в непосредственной близости от «истинного» гена болезни [Elston R., 1998; Thomson G., Esposito M, 1999]. Независимо от того, какая из двух возможных причин генетической ассоциации имеет место при конкретном заболевании, выявление такой ассоциации является важнейшим шагом в уточнении молекулярных механизмов наследственной предрасположенности к данному заболеванию: в первом случае речь идет непосредственно об идентификации гена предрасположенности, во втором - об установлении хромосомной локализации такого гена.
При анализе генетических ассоциаций могут использоваться как выборки изолированных случаев заболевания, так и группы семей, в которых имеет место накопление повторных случаев данного заболевания. Клю
чевым требованием является одинаковый этнический состав группы больных и контрольной группы, поскольку частоты конкретных аллелей изучаемых генетических маркеров могут существенно различаться в разных популяциях [Thomson G., 1995 (б); Thomson G., Esposito М., 1999]. Еще одним важным условием при изучении генетических ассоциаций является знание (хотя бы в общих чертах) некоторых звеньев патогенеза изучаемого муль- тифакториального заболевания, поскольку на основании этого может быть отобран круг соответствующих генов- кандидатов для проверки теоретической гипотезы об их возможной роли в формировании предрасположенности к данному заболеванию.
В неврологии классическим примером выраженной генетической ассоциации является значение аполи- попротеина Е как важнейшего эндогенного фактора риска в развитии поздней формы болезни Альцгеймера - заболевания, которым в мире страдают около 20 млн. человек. Аполипопротеин Е (апоЕ) представляет собой белок с молекулярной массой 34 кДа, кодируемый геном на хромосоме 19ql3.2 [Mahley R., 1988]. АпоЕ играет ключевую роль в метаболизме липидов (особенно холестерина), способствуя их перераспределению между клетками различных органов [Mahley R., 1988; Mahley R. et al., 1996]. В 1993 году W. Strittmatter с соавторами установили, что апоЕ является одним из протеинов, специфически связывающихся с Р-амилоидом (основным компонентом амилоидной бляшки - морфологического маркера болезни Альцгеймера); это послужило основанием для детального анализа ассоциации апоЕ и болезни Альцгеймера. Известно, что ген апоЕ имеет 3 основных аллеля (s2, еЗ и е4), отличающихся единичными нуклеотидными заменами и определяющих существование 3 изоформ белка апоЕ, причем в общей популяции аллель еЗ наиболее раснространен. В серии исследований, проведенных в 1993-1996 гг., было установлено, что аллель s4 гена апоЕ встречается достоверно чаще у больных с поздней формой болезни Альцгеймера - как семейной (50%), так и спорадической (40%), в то время как в контрольной группе его частота не превышает 14-16% [Strittmatter W. et al., 1993; Saunders A. et al., 1993; Locke P. et al., 1995; Schellenberg G., 1995; Strittmatter W., Roses A., 1996]. Более того, риск развития на протяжении жизни болезни Альцгеймера в зависимости от генотипа апоЕ является доза-зависигмым: у гомозиготных носителей аллеля s4 он является наивысшим и составляет около 90% (т.е. 90% лиц с генотипом s4/s4 заболеЕОт при условии достижения возраста ~70- 75 лет), у гетерозиготных носителей s4 он равен 47%, тогда как лишь 20% лиц, не имеющих аллеля s4, заболеют болезнью Альцгеймера в пожилом возрасте [Corder Е et al., 1993; van Broeckhoven С., 1995; Blacker D., Tanzi R., 1998]. Доза «неблагоприятного» аллеля s4 напрямую коррелирует также с интенсивностью формирования амилоидных бляшек в мозге больных с болезнью Аль- цеймера [Schmechel D. et al., 1993]. Интересно отметить, что средний возраст начала заболевания у гомозиготных носителей аллеля е4 является достоверно более ранним (68 лет) по сравнению с гетерозиготными носителями апоЕ-е4 (75 лет) и лицами с другими генотипами апоЕ (84 года). Другими словами, каждая копия аллеля е4 «приближает» возраст начала болезни Альцгеймера на 5-9 лет [Corder Е. et al., 1993; Blacker D., Tanzi R., 1998]. С другой стороны, носительство аллеля s2 (особенно двух его копий) обладает протективным эффектом в отношении развития на протяжении жизни болезни Альцгеймера [Corder Е. et al., 1994]. Подтверждением патогенетической значимости ассоциаций аллелей апоЕ и болезни Альцгеймера являются данные о взаимосвязи различных генетических вариантов алоЕ с особенностями клинической картины заболевания и реакции на проводимое лечение [Lovestone S., 1999]. Указанные закономерности действия различных вариантов гена апоЕ в качестве мощных факторов риска и антириска болезни Альцгеймера справедливы практически для всех исследованных популяций мира [Schellenberg G., 1995].
Открытие роли гена апв{ в развитии болезни Альцгеймера в позднем возрасте поставило вопрос о клиническом значении обнаружения у конкретных индивидуумов «критического» аллеля anoE-s4. Как известно, в целом для пожилых людей риск развития болезни Альцгеймера сам по себе является достаточно высоким и повышается с каждым прожитым годом. Так, распространенность болезни в группе лиц 65-74 года составляет 3%, для следующего десятилетия - 18% и для лиц старше 85 лет - около 47% [Katzman R., 1976]; следовательно, в связи с неуклонным постарением населения все большее число пожилых людей будет попадать в категорию относительного высокого риска по болезни Альцгеймера. Таким образом, изменение степени этого риска в зависимости от наличия конкретных аллелей апоЕ не носит «драматический» характер и, по-видимому, не является в настоящее время основанием для проведения широкомасштабных программ генетического тестирования пожилого населения, направленных 1на установление вероятности развития у них болезни Альцгеймера [Seshardi S. et al., 1995; National Institute on Aging/ Alzheimer’s Association Working Group, 1996; Lovestone S., 1999]. Такое тестирование, по-видимому, могло бы основываться на комбинированном исследовании гена апоЕ и дру- |-и:х полиморфных генов, определенные варианты которых привносят дополнительный риск в отношении развития болезни Альцгеймера [Roses А., 1997; Sheu К. et al., 1999; Zubenko G. et al., 1999]. В будущем, при внедрении в практику высокоэффективных методов лечения болезни Альцгеймера, прогностическое ДНК-тестиро- вание пожилых лиц с выявлением носительства anoE-s4 и других «неблагоприятных» аллелей может принять более активный и целенаправленный характер, связанный с необходимостью формирования достоверной группы «высокого риска» и возможностью проведения таким клинически здоровым людям превентивной терапии. Несколько иначе обстоит дело с ДНК-диагности- кой аллельных вариантов гена апоЕ у пожилых больных, обратившихся в клинику с нарушениями памяти и другими симптомами, позволяющими предполагать у них раннюю стадию болезни Альцгеймера. Большинством авторов признается, что при наличии сложностей в клинической диагностике обнаружение аллеля s4 (и особенно генотипа s4/s4) может, наряду с результатами других методов исследования, служить важным дополнительным диагностическим маркером, косвенно подтверждающим диагноз болезни Альцгеймера [Saunders A. et al., 1996; Lovestone S., 1999]. Окончательное решение этого вопроса требует дальнейшего накопления данных (в особенности - проспективного наблюдения за тестированными лицами и анализа репрезентативных серий секционных случаев), поэтому в настоящее время практическая роль ДНК-тестирования гена апоЕ с целью ранней диагностики болезни Альцгеймера остается дискуссионной [Blacker D., Tanzi R., 1998].
Таким образом, в клинической медицине болезнь Альцгеймера является первым примером распространенного заболевания, для которого установлен ведущий генетический фактор предрасположенности. В связи с этим болежь Альцгеймера может рассматриваться как
своеобразная модель для разработки методологических аспектов ДНК-тестирования при мультифакториальных болезнях человека (включая решение медицинских, социально-правовых и этических вопросов) [Roses А., 1997]. Если при наследственных моногенных болезнях мутационный анализ касается сравнительно немногочисленной группы риска в отягощенных семьях, то при болезни Альцгеймера результаты ДНК-тестирования, потенциально несущие благоприятный или неблагоприятный прогноз, затрагивает десятки миллионов людей. Именно поэтому, как указывалось выше, внедрение в практику методов ДПК-анализа апоЕ и других генов предрасположенности к болезни Альцгеймера не может форсироваться и должно основываться только на строго доказанных и многократно подтвержденных положениях. На сегодняшний день очевидно, что сам по себе ген апоЕ не является единственным детерминантом «генетической судьбы» обследуемых лиц, что подтверждается следующими хорошо известными фактами: а) не менее чем в половине случаев болезнь Альцгеймера развивается у людей, не имеющих «неблагоприятного» аллеля anoE-s4; б) у носителей аллеля е4 риск развития болезни Альцгеймера, хотя и значительно превышает об- щепопуляционный, но все же не является абсолютным; 3) отмеченная выше ассоциация anoE-s4 и болезни Альцгеймера заметно снижается в популяции наиболее пожилых лиц, переживших «критический» возраст (gt;80 лет) [van Broeckhoven С., 1995; Roses А., 1997; Sheu К. et al., 1999]. Обобщая сказанное, можно еще раз подчеркнуть, что ген апоЕ является лишь одним из звеньев сложной «мозаики» взаимодействующих факторов, определяющих возможность развития позднего варианта болезни Альцгеймера.
Молекулярные механизмы, объясняющие повышенную предрасположенность к болезни Альцгеймера у носителей аллеля апоЕ-е4, были предметом интенсивных исследований последних лет. Основное внимание при этом уделялось изучению кинетики межмолекулярных взаимодействий различных изоформ белка апоЕ, каждой из которых свойственны свои особенности конформации пептидной молекулы. Имеющиеся в литературе данные позволяют предполагать, что в4-изоформа белка характеризуется более выраженным амилоидогенным потенциалом благодаря высокому сродству к [3-ами- лоиДу, а также относительно непрочным связыванием с белком тау (гиперфосфорилирование свободных молекул которого лежит в основе образования характерных для болезни Альцгеймера нейрофибриллярных клубков) [Schmechel D. et al., 1993; Weisgraber К. et al., 1994; Schelleberg G., 1995; van Broeckhoven C., 1995]. Эксперименты на трансгенных животных свидетельствуют о прямом участии апоЕ в формировании фибриллярных депозитов Р-амилоида [Mattson М., 1997]. Обсуждается также роль апоЕ как сигнального протеина, регулирующего механизмы липидного транспорта (в частности, мобилизации «строительного» холестерина) в процессе синаптогенеза и регенерации нейрональных мембран [Mahley R. et al., 1996]. Последняя гипотеза подтверждается значительным (в несколько сот раз) повышением уровня экспрессии апоЕ при эксперименгальном повреждении седалищного нерва [Ignatius М. et al., 1986]. Е1аи- более вероятным представляется комплексный характер участия апоЕ в патогенезе болезни Альцгеймера.
Предположение об универсальной роли апоЕ в механизмах повреждения и регенерации нейронов обусловило многочисленные исследования, направленные на анализ генетических ассоциаций между конкретными аллелями апоЕ и рядом других мультифакториальных заболеваний центральной нервной системы (инсульт, болезнь Паркинсона, множественные системные атрофии и др.). В этих исследованиях наиболее определенные результаты были достигнуты при изучении группы нейро- дегенеративных заболеваний. J. Schneider с соавторами (1995) обнаружили повышенную частоту аллеля е4 у больных кортико-базальной дегенерацией, болезнью Пика и прогрессирующим сунрануклеарным параличом. В нескольких недавних исследованиях было показано, что у больных болезнью Паркинсона ген апоЕ модулирует возраст начала заболевания, поскольку носительство аллеля s4 было достоверно ассоциировано с более ранним дебютом симптомов (на4-7 лет) [Zareparsi S. etal., 1997; Kruger R. et al., 1999]. В то же время, распределение различных аллелей апоЕ в группе больных болезнью Паркинсона не отличается от такового в общей популяции [Rubinsztein
- et al.. 1994; Roller W. et al., 1995]. Наши предварительные данные, полученные при изучении спорадической болезни Паркинсона в российской популяции, также свидетельствуют в пользу более раннего начала заболевания у носителей «неблагоприятного» аллеля s4, что может служить подтверждением роли апоЕ в механизмах предрасположенности к данному заболеванию. На рис. 71 показан пример тестирования гена апоЕ у больных болезнью Паркинсона.
При болезни Паркинсона проводился анализ ассоциаций с целым рядом других генов-кандидатов, которые, предположительно, могут быть связаны с различными звеньями патогенеза данного заболевания. В числе таких кандидатов исследовались гены дофаминовых рецепторов, переносчиков дофамина, моноаминоксидаз, I юкоторых ферментов, метаболизирующих разнообразные Фзотоксины или участвующих в реакциях окислительного фосфорилирования, митохондриальные гены и др. (таб- пнца 8).
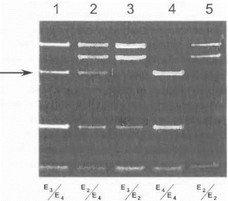
Рис.71. Анализ полиморфизма в гене апоЕ при болезни Паркинсона
Электрофореграмма продуктов рестрикции (использована рестрик- таза Hha I). Дорожки I 4 - больные с раипсп формой болезни Паркинсона, дорожка 5 больной с поздней формой болезни Паркинсона. Внизу под каждой дорожкой обозначены генотипы больных. Стрелкой указан фрагмент длиной 72 п.о., специфичный ЙЛя аллеля е4. Три из четырех обследованных больных с ранней формой болезни Паркинсона являются носителями аллеля е4 в гомо- или гетерозиготном состоянии.
Проведенные исследования показали, что в отдельных выборках больных болезнью Паркинсона наблюдается изменение распределения аллельных частот генов дофаминовых 02-рецепторов, моноаминоксидаз А- и В- типов, тирозин-гидроксилазы, N-ацетилтрансфера- зы, 4-гидроксилазы цитохрома Р450 (CYP2D6), Е2-субъе диницы а-кетоглутарат-дегидрогеназного комплекса и некоторых митохондриальных полиморфизмов [Shoffner J. etal., 1993;HiguchiS.etal., 1995;Nussbaum R., Polymeropoulos M., 1997; WoodN., 1997; Kobayashi T. el al., 1998;Nicholl D. etal., 1999; Spacey S., WoodN., 1999].
Таблица 8
Анализ генетических ассоциаций
при болезни Паркинсона
|
Исследованный ген-кандидат (белок) |
Результат анализа |
|
Аполипопротеин Е(апоЕ) |
+ |
|
Дофаминовый D2-pcuenrop (DRD2) |
+ - |
|
Дофаминовый йЗ-рецсптор (DRD3) |
- |
|
Дофаминовый 04-рецсптор (DRD4) |
- |
|
Транспортер дофамина (DAT 1) |
- |
|
Моноаминоксидаза А-типа (МАО-А) |
+ - |
|
Моноаминоксидаза В-типа (МАО-В) |
+ - |
|
Тирозин-гидроксилаза |
+ — |
|
Е2-субъединица а-кстоглутарат-дегидрогемазного комплекса |
+ |
|
Семейство генов цитохрома Р450 (CYP1AI, CYP2D6, CYP2E1] |
+ - |
|
Глутатион-гран сфер азы Ml и TL |
- |
|
N-ацетилтрансфераза 2 (NAT2) |
- |
|
NAD PH-редуктаза |
- |
|
Митохондриальные полиморфизмы (нуклеотидные положения 3397, 4336, 5460 и др.) |
+ - |
Примечание: означает подтвержденную ассоциацию (повышение частоты
определенного аллеля гена у больных болезнью Паркинсона или модулирующий эффект аллеля на возраст начала заболевания), “+ означает ассоциацию, выявленную лишь в отдельных выборках обследованных больных, означает отсутствие ассоциации.
Однако указанные генетические ассоциации при болезни Паркинсона не во всех случаях могут быть подтверждены при исследовании других популяций, что может отражать существование определенных межпопуляцион- пых различий аллельных частот изучаемых генов, а также различий удельного веса разнообразных генетических и средовых факторов в патогенезе болезни Паркинсона [Spacey S., Wood N., 1999]. Таким образом, в механизмах предрасположенности к болезни Паркинсона важную роль играет совокупное действие ряда генов, контролирующих активность дофаминергической трансмиссии, процессы детоксикации и клеточной энергетики. Вклад этих генетических факторов, детерминирующих определенный «метаболический фон» у близких родственников, а также сходный характер разнообразных экзогенных воздействий у членов одной семьи лежат в основе нередко наблюдающегося накопления повторных случаев болезни Паркинсона в обследуемых семьях; не случайно положительный семейный анамнез по болезни Паркинсона выявляется у 10,3% больных с этим заболеванием (против 3,5% в контрольной группе) [ElbazA. etal., 1999].
Прогрессирующий супрануклеарный паралич (ПСП) представляет собой второй по частоте (после болезни Паркинсона) синдром паркинсонизма дегенеративной природы [Bowel J. et al., 1997]. В абсолютном большинстве случаев ПСП является спорадическим заболеванием, имеющим мультифакториальную природу, поэтому в последние годы основное внимание было сфокусировано на поиске генов предрасположенности к данному тяжелому страданию. Характерным нейрогистохи- мическим критерием ПСП является формирование линейных нейрофибриллярных филаментов, образованных преципитатами гиперфосфорилированной формы клеточного белка тау [Bergeron С. et al., 1998]. В связи с этим рядом авторов исследовалась генетическая структура тау у больных ПСП. Показано, что в группе больных данным заболеванием достоверно чаще встречается сочетание ряда специфических полиморфных участков гена тау, образующих характерный редкий гаплотип, практически не наблюдающийся у лиц контрольной группы [Baker М. et al., 1999; Higgins J. et al., 1999]. Данный гаплотип сопровождается экспрессией в веществе мозга (особенно в стволе - первичной «мишени» патологического процесса при ПСП) особой изоформы белка тау, взаимодействие которой с микротубулярным аппаратом нейронов и клеток глии существенно меняется [Chambers С. et al., 1999]. Предполагается, что такой механизм способствует образованию нейрофибриллярных преципитатов, являющихся важнейшим звеном патогенеза ПСП. Таким образом, доказанная ассоциация ПСП с определенными генетическими вариантами тау, а также (как указывалось выше) с аллелем е4 гена апоЕ позволяет говорить об идентификации, как минимум, 2 генов предрасположенности к развитию ПСП. Для уточнения диагностического и прогностического значения результатов ДНК-тестирования указанных генов в группе риска (например, у родственников больных ПСП) требует ся дальнейший анализ катамнестических данных на основе многолетних наблюдений за обследованными группами клинически здоровых лиц.