
Митохондриальные энцефаломиопатии представляют собой группу заболеваний, относящихся к особому классу наследственной патологии человека - митохондриальным болезням (митохондриальным цитопатиям). В самом общем виде митохондриальные болезни можно определить как заболевания, обусловленные генетическими и структурно-биохимическими дефектами митохондрий и сопровождающиеся нарушением тканевого дыхания [Вельтищев Ю.Е., Темин П.А., 1998; Крас- попольская К.Д., Захарова Е.Ю., 1998; Shoffner Г, Wallace D., 1992; DiMauro S., 1993].
Митохондрия - это внутриклеточная органелла, играющая центральную роль в энергетическом метаболизме клетки. Функция митохондрий заключается в
ступенчатом высвобождении химической энергии окисления органических субстратов и ее превращении в энергию макроэргических фосфатов. Таким образом, в митохондриях обе стороны двуединого процесса - аэробное окисление и окислительное фосфорилирование - являются сопряженными. Аэробное окисление крупных органических молекул реализуется через образование более простых органических соединений (глюкоза, жирные кислоты, глицерол, аминокислоты) и их дальнейшее окисление до специфических промежуточных продуктов (ацетил-КоА), вступающих в цикл трикарбоно- вых кислот Кребса. В цикле Кребса реализуется серия окислительно-восстановительных реакций, в которых атомы водорода акцептируются адениновыми и флави- новыми нуклеотидами (НАД+ и ФАД’1'). Финальной стадией является окисление восстановленных форм НАДН и ФАДН2 в так называемой дыхательной цепи митохондрий - системе из 5 ферментативных комплексов, последовательно транспортирующих электроны и протоны на кислород с образованием молекул воды. Энергия, высвобождающаяся в процессе переноса электронов, накапливается в виде макроэргических фосфатов (АТФ и др.). Основные химические реакции происходят на внутренней мембране митохондрий, имеющей значительную площадь благодаря многочисленным складкам (кристам), инвагинирующим в митохондриальный матрикс. Помимо участия в энергетическом метаболизме, митохондрии выполняют также ряд дополнительных важных функций - таких как синтез аминокислот, ииримидинов, липидов, гема и других метаболитов.
Сложность и многообразие митохондриальных функций находят свое прямое отражение на белковом уровне: в митохондриях локализовано около 1000 различных полипептидов, что составляет почти 10% всего белкового пула типичной эукариотической клетки. Уникальной особеностью митохондрий является тот факт, (что источником митохондриальных белков являются 2 различных самостоятельных генома клетки — ядерный и собственно митохондриальный. Абсолютное большинство белков, принимающих участие в функционировании митохондрий, синтезируются в ядре клетки и впос- педствии транспортируются к соответствующему митохондриальному компартменту. В то же время 13 полипептидов (все из них являются компонентами дыхательной цепи) синтезируются генами, входящими в состав митохондриальной ДНК.
Митохондриальная (внеядерная, цитоплазматическая) ДНК представляет собой автономную по отношению к ядру генетическую систему, организованную в виде двуцепочечной кольцевой молекулы (рис. 67). Митохондриальную ДНК (мтДНК) иногда условно называют М-хромосомой. Структура мтДНК полностью расшифрована: молекула мтДНК состоит всего из 16569 и.о. и содержит 37 генов. Эти гены кодируют синтез 2 видов рибосомальной РНК, 22 видов транспортной РНК (необходимых для синтеза белка в митохондриях), а также 13 белков, входящих в состав I, III, IV и V комплексов дыхательной цепи митохондрий.
Значительная часть белков этих комплексов, а также все белки II комплекса дыхательной цепи, кодируются ядерпой ДНК. Гены мтДНК расположены очень компактно, не прерываясь интронными вставками и нс образовывая комплексов с белками-гистонами. Отсутствие «гистонной защиты», а также относительное несовершенство системы репарации и высокая подверженность воздействию свободных радикалов, образуемых при аэробном окислении, способствуют весьма высокой скорости накопления мутаций в мтДНК в онто- и филогенезе. Повышенный темп мутирования мтДНК обус-
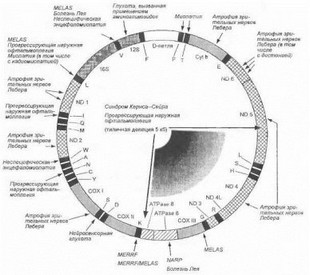
Рис. 67. Митохондриальная ДНК человека и основные мутации при митохондриальных энцефаломиопатиях Показано подразделение кольцевидной молекулы мтДНК на отдельные гены. Гены мтДНК указаны вдоль внутренней поверхности кольца:
- символы ND 1 - ND 6 и ND 4L обозначают семь субъединиц комплекса I дыхательной цепи (NADH-ubiquinone oxidoreductase);
- символ Cyt b обозначает субъединицу комплекса III (ubiquinoi- cytochrome с oxidase oxidoreductase);
- символы СОХ I - СОХ III обозначают три субъединицы комплекса IV (cytochrome с oxidase);
- символы ATPase 6 и 8 обозначают две субъединицы комплекса V (АТР synthase);
- символы 16S и 12S обозначают гены рибосомальных РНК;
- однобуквепные символы обозначают гены транспортных РНК (в соответствии с однобуквенными символами 22 транспортируемых аминокислот, указанными в приложении 2).
В центре кольца указаны границы типичной делеции протяженностью 5 кб, обусловливающей развитие синдрома Ксрнса-Сейра и прогрессирующей наружной офтальмоплегии. Наиболее частые точковые мутации, обусловливающие развитие митохондриальных энцефаломиопатий, обозначены вдоль наружной поверхности кольца.
ловливает высокую частоту спорадических случаев митохондриальных болезней.
Число митохондрий в клетках различных тканей может составлять от сотен до нескольких тысяч, а каждая митохондрия содержит от 2 до 10 копий мтДНК. Общее же количество молекул мтДНК в клетке может достигать десятков тысяч. Например, в одном кардиомиоците может содержаться до 50 000 молекул мтДНК. Каждая молекула мтДНК реплицируется самостоятельно, деление митохондрий в цитоплазме также представляет собой автономный процесс. В результате клеточно- ] о деления (митоз, мейоз) различные молекулы мтДНК в составе митохондрий в случайном порядке переходят в цитоплазму дочерних клеток. В обычной ситуации во всех клетках и тканях организма имеется один и тот же нормальный вид мтДНК - состояние, обозначаемое как гомоплазмия. При возникновении мутации в мтДНК и дальнейшей амлификации мутантного клона возникает 1 етероплазмня,*г.е. состояние, при котором в клетке (ткани) существует совокупность двух различных популяций мтДНК - нормальной и мутантной. Процентное содержание нормальной и мутантной мтДНК в конкретной гкаии может варьировать в широких пределах (от 0 до 100%), что в значительной степени определяет тяжесть соответствующих клинических проявлений болезпи. Поскольку репликация митохондрий и мтДНК является автономной по отношению к клеточному циклу, а их распределение по дочерним клеткам имеет случайный характер, в организме имеет место митотическая сегрегация мтДНК, проявляющаяся постоянным динамическим изменением соотношения нормального и мутан- .ного пулов мтДНК по мере обновления клеточных популяций. Следовательно, уровень гетероплазмии является различным как в разных тканях, так и в одной и той же ткани в различные периоды жизни. В определенных тканях митотическая сегрегация мтДНК может приводить к полному замещению нормальных молекул мтДНК мутантными, с переходом от состояния гетероплазмии к состоянию гомоплазмии по мутантной мтДНК. Таким образом, по образному выражению A. Schon и S. DiMauro (1994), по своей сути вся митохондриальная генетика является популяционной генетикой, основанной на закономерностях динамики соотношения между нормальной и мутантной популяциями мтДНК. Данные закономерности имеют большое значение для понимания клинических особенностей и методов молекулярной диагностики митохондриальных болезней (см. далее).
Тяжесть поражения отдельных тканей и органов при митохондриальных болезнях определяются не только характером генетического дефекта мтДНК и уровнем гетероплазмии, но и их потребностью в энергии и зависимостью клеточных функций от эффективности окислительного фосфорилирования. Наиболее энергозависимыми являются ЦНС, скелетная и сердечная мускулатура, глаз, почки, эндокринные железы, поэтому именно эти ткани наиболее часто и закономерно вовлекаются в патологический процесс при митохондриальных дефектах. Для манифестации симптоматики со стороны того или иного органа или ткани необходимо, чтобы митохондриальная дисфункция и нарушение окислительного фосфорилирования в них превысит определенный критический порог, специфический для каждой ткани. Например, для ЦНС таким пороговым значением является содержание мутантного вида мтДНК в нейронах около 60%; если у больных содержание мутантной мтДНК в ЦНС превышает эту цифру, в клинической картине развиваются неврологические симптомы. Для других тканей порог переносимости митохондриальной дисфункции выше. Тяжесть митохондриальных болезней с течением времени неуклонно нарастает, что обусловлено возрастным снижением активности процессов окислительного фосфорилирования и накоплением мутаций мтДНК в стареющих тканях.
Митохондриальные болезни могут наследоваться по различным типам, что зависит от характера генетического дефекта при конкретном заболевании. Поскольку вся мтДНК в организме имеет материнское происхождение (из цитоплазмы яйцеклетки), в случае передачи митохондриальной мутации потомству в родословной имеет место материнский тип наследования - когда заболевание проявляется у всех детей больной матери. Если мутация происходит в ядерном гене, кодирующем синтез митохондриального белка, заболевание передается но классическим менделевским законам (чаще всего по аутосомно-доминантному и аgt;тосомно-рецессив- ному типам). Наконец, при ряде форм митохондриальных болезней мутация мтДНК возникает de novo на ранней стадии онтогенеза, приводя в результате митотической сегрегации к преимущественному накоплению мутантного вида мтДНК в тканях-мишенях; в такой ситуации имеет место спорадический случай болезни.
Рассматриваемые в настоящей главе митохондриальные энцефаломиопатии представляют собой частную группу митохондриальных болезней, в клинической картине которых превалирует поражение мозга и поперечно-полосатой мускулатуры.