
Профилактика наследственной патологии и уменьшение «генетического груза» в популяции представляет собой одну из важнейших медицинских и социально-экономических задач, которые стоят перед современным индустриальным обществом, испытывающим беспрецедентное давление неблагоприятных экологических факторов. Эта задача является тем более актуальной, когда речь идет о наследственных заболеваниях нервной системы, характеризующихся в большинстве случаев тяжелой физической и психической инва- лидизацией больных. Профилактика наследственных заболеваний может быть первичной (предупреждение зачатия или рождения больного индивида) и вторичной (разнообразные способы коррекции патологического генотипа) [Бочков Н.П., 1997]. На современном этапе развития клинической нейрогенетики возможности вто
ричной профилактики наследственной патологии (т.е. патогенетического лечения нейрогередитарных заболеваний) достаточно ограниченны, поэтому на первый план выходит предотвращение появления «генетически больного» потомства. Этой цели служит медико-генетическое консультирование - особый вид специализированной медицинской помощи, направленный на предупреждение появления повторных случаев наследственных заболеваний в отягощенных семьях.
Медико-генетическое консультирование включает несколько основных задач [Козлова С.И. и др., 1996; Бочков Н.П., 1997; Доклад научной группы ВОЗ, 1997]:
а) установление точного диагноза и типа наследования заболевания в консультируемой семье;
б) расчет генетического риска у консультируемых; родственников, включая (при возможности) точное установление их генетического статуса с помощью методов ДНК- диагностики {прогностическое тестирование);
в) определение прогноза потомства и определение наиболее эффективного способа профилактики новых случаев заболевания (в том числе с помощью пренатальной ДНК-диагностики плода на ранних сроках беременности);
г) объяснение консультируемым лицам смысла полученной и проанализированной информации, решение возникающих юридических и морально-этических проблем;
д) помощь консультируемой семье в решении целого ряда других вопросов, касающихся планирования жизни, репродуктивного поведения и возможности деторождения, психологической поддержки, социальной адаптации и т.д.
Точная диагностика наследственного заболевания нервной системы и установление типа наследования патологического фенотипа являются первым необходимым этапом медико-генетического консультирования обратившегося за помощью лица (семьи). Этот этап может осуществляться, в зависимости от конкретных медико-организационных условий, врачом общей практики, неврологом, врачом-генетиком медико-генетической консультации либо (в идеале) квалифицированным специалистом в области клинической нейрогенетики. Особенности генеалогического анализа в неврологии и постановки диагноза наследственного заболевания нервной системы подробно проанализированы в главе 1 (раздел 1.2) и в соответствующих частных разделах главы 3. Хотелось бы еще раз подчеркнуть значение детального обследования всех членов консультируемой семьи, а также необходимость выявления у родственников любых скрытых проявлений носительства мутантного гена, в том числе с использованием комплекса дополнительных лабораторно-инструментальных методов исследования.
Адекватно проведенный клинико-генеалогический анализ позволяет выделить в консультируемой семье так называемую группу риска, которая и является основным объектом мсдийо-генетического консультирования. Под группой риска понимаются родственники пробанда, которые в соответствии с типом наследования могут являться носителями мутантного гена и имеют высокий риск заболеть данным наследственным заболеванием или передать мутантный ген потомству. Например, при хорее Гентингтона, характеризующейся аутосомно-доминантным типом наследования, практически полной пенетрантностью и поздним возрастом начала болезни, к группе риска следует относить детей пробанда, а также его братьев-сестер: для каждого из этих лиц при рождении теоретический риск унаследо-
Таблица 9
Расчетные значения вероятностей фенотипов потомства при известных типах брака (для менделирующих заболеваний)
Примечание: вероятности фенотипов в таблице представлены для случаев с
полной пенегрантностью мутантного гена А-доминантный аллель (мутантный в случае юминантных заболеваний), а — рецессивный аллель (мутантный в случае рецессивных заболеваний).
вать мутацию и заболевание составляет 50%, но при этом для здоровых лиц риск заболеть постепенно снижается с каждым прожитым десятилетием (особенно после прохождения наиболее «опасного» возраста в 40-60 лет).
При болезни Фридрейха, наследующейся по аутосом- но-рецессивному типу и начинающейся обычно на втором десятилетии жизни, основную группу риска составляют братья и сестры пробанда (теоретический риск 25%), причем степень риска заболевания выше у младших сибсов, не достигших «критического» возраста манифестации болезни. При Х-сцепленной мышечной дистрофии Дюшенна/Бекера к группе риска по заболеванию относятся лица мужского пола - родственники пробанда по материнской линии; помимо этого, сестры пробанда также имеют высокий (50%) риск унаследовать от матери мутантный ген и, оставаясь клинически здоровыми, передать заболевание мужчинам в следующее поколение.
Как видно из представленных примеров, выделение группы риска неразрывно связано с определением у данной категории лиц генетического риска - т.е. вероятности появления у них или их потомства соответствующего наследственного заболевания. Генетический риск до 5% оценивается как низкий и не считается противопоказанием к деторождению в консультируемой семье. Риск от 6 до 20% принято считать средним, а свыше 20% - высоким. При среднем и высоком риске рекомендации относительно возможности деторождения определяются в основном тяжестью медицинских и социальных последствий конкретного заболевания, а также возможностями и доступностью пренатальной ДНК-диагностики [Козлова С.И. и др., 1996].
До использования методов ДНК-диагностики генетический риск при менделирующих заболеваниях определяется путем вероятностных расчетов, основанных на закономерностях сегрегации патологического фенотипа при том или ином типе наследования. Точность расчета генетического риска зависит от информативности родословной и возможности установления генотипов консультируемых родителей. Если генотипы родителей (тип брака) известны или могут быть предположены с высокой вероятностью, оценка риска манифестации патологического фенотипа у потомков сравнительно проста - см. таблицу 9 [Козлова С.И. и др., 1996]. В необходимых случаях делается поправка на неполную пенетрантность мутантного гена, коэффициент инбридинга и некоторые другие факторы, влияющие на проявление фенотипа. Значительно сложнее расчет генетического риска при неизвестных генотипах родителей. Обычно это имеет место при наличии единичных случаев заболевания в семье, особенно если изучаемый синдром является генетически гетерогенным: в такой ситуации оценка сегрегации признаков в родословной неосуществима, и расчет генетического риска приходится проводить для различных возможных типов наследования. В этих случаях используются специальные математические формулы, оспованные на теории вероятности. Данные формулы при расчете общего риска принимают во внимание априорные факторы (менделевские закономерности предполагаемого типа наследования и сравнительную частоту возможных генетических вариантов болезни), а также фактическое наличие у консультируемого лица больных или здоровых родственников (что определяет апостериорную вероятность рассматриваемой гипотезы). При неизвестном типе брака рождение каждого последующего ребенка (здорового или больного) существенно дополняет имеющуюся генетическую информацию и видоизменяет величину апостериорной вероятности.
Нри мультифакториальных заболеваниях с поли- генно-детерминируемой наследственной предрасположенностью суммарный генетический риск зависит от большого числа факторов и определяется с помощью специальных таблиц эмпирического риска.
Возможность ДНК-диагностики существенно повышает точность определения генетического риска, позволяя от вероятностных моделей перейти к однозначному (дефинитивному) определению генотипов и ожидаемых фенотипов. В практике медико-генетического консультирования методы ДНК-анализа могут использоваться для пренатальной, преимплантационной и ранней доклинической диагностики носительства мутации. Пренатальная Д] IK-диагностика предполагает определение генотипа плода на ранних сроках беременности (пер- вый-второй триместр, предпочтительнее на 8-12-й неделе), что дает возможность при обнаружении мутации сделать медицинский аборт. Образцы ДПК плода получают из биоптатов хориона (хорионбиопсия), плаценты (плацентобиопсия), клеток амниотической жидкости (ам- ниоцентез) или лимфоцитов пуповинной крови (кордо- центез). К сожалению, все указанные процедуры, проводящиеся под контролем ультразвука, являются инвазивными и сопряжены с определенным риском прерывания беременности, оцениваемым в среднем как 1,5-2%. Новым неинвазивным методом в пренатальной диагностике является анализ ядросодержащих клеток плода, присутствующих в крови матери [Золотухина Т.В., Шилова Н.В., 1999]. Эти клетки (трофобластные клетки, лимфоциты, гранулоциты, эритробласты плода) .появляются в организме матери в результате трансплацентарного переноса, способствуя развитию толерантности материнского организма по отношению к плоду; их концентрация составляет, ориентировочно, 10-5—10 s. В настоящее время наиболее серьезной проблемой является недостаточная эффективность существующих методов сортировки и детекции плодных клеток, получаемых из крови матери. Одним из возможных решений данной проблемы является многократная амплификация генома единичных клеток плода с помощью полимеразной цепной реакции. По-видимому, перспективы более широкого использования анализа клеток плода из крови матери с целью пренатальной ДНК-диагностики связаны с дальнейшим развитием методов обогащения популяции плодных клеток.
Если в обследуемой семье мутация изучаемого гена известна (т.е. выявлена ранее у больного родственника или клинически здорового носителя), при пренатальной ДНК-диагностике плода молекулярное тестирование облегчается - например, путем использования соответствующей рестрикционной эндонуклеазы. В том случае, когда предварительный генетический материал больного члена семьи (носителя) недоступен для исследования, поиск мутаций у плода осуществляется стандартными методами мутационного скрининга - в зависимости от генетических особенностей изучаемого заболевания, подробно разбираемых в главе 3. Выявление конкретных мутаций у плода позволяет с абсолютной достоверностью устанавливать его генетический статус и определять прогноз. Несколько иначе обстоит дело с пренатальной ДНК-диагностикой, осуществляемой косвенными методами. Такая диагностика обычно проводится в тех случаях, когда изучаемый ген не идентифицирован (известна лишь его хромосомная локализация) либо он имеет сложную структуру, что технически значительно затрудняет поиск мутаций. Точность косвенной ДНК-диагностики, позволяющей оценивать наследование мутантной хромосомы, определяется близостью патологического гена и используемого маркера (маркеров). Например, если ген болезни и изучаемый маркер
расположены друг от друга на расстоянии 2 сМ, вероятность рекомбинации между данными локусами (и следовательно, вероятность ошибки) составляет 2%; соответственно, точность диагностики носительства мутантной или нормальной хромосомы в этом случае составляет около 98%. Идеальным является использование внутригенных маркеров в комбинации с тесно сцепленными маркерами, фланкирующими изучаемый ген с теломерной и центромерной сторон.
Весьма перспективным направлением профилактики наследственной патологии является преимпланта- ционная ДНК-диагностика. Сущность ее заключается в исследовании ДНК клеток раннего зародыша, развитие которого инициируется in vitro путем искусственного оплодотворения (альтернативой является анализ ДНК полярных телец овулировавших яйцеклеток). Имплантация зародыша в матку осуществляется только после исключения у него носительства мутантного гена. Таким образом исход соответствующей беременности становится, с генетической точки зрения, однозначно благоприятным. В неврологии наибольший опыт преимп- лантационной ДНК-диагностики получен при мышечной дистрофии Дюшенна/Векера, спинальной амиотрофии и некоторых других моногенных заболеваниях (Vertinsky Y., Kuliev А., 1993; Fallon L. etal., 1999]. До настоящего времени основной проблемой преимплантационной ДНК- диагностики является недостаточно высокий процент приживаемости имплантируемых в матку зародышей (не более 20-30%). Однако с развитием микрохирургической техники и общим прогрессом гинекологии и эндокринологии можно ожидать новых существенных успехов в техническом и медико-биологическом обеспечении преимплантационной диагностики наследственных заболеваний.
Методы ДНК-диагностики носительства мутаций у клинически здоровых лиц, принадлежащих к группе риска, принципиально не отличаются от аналогичных подходов, используемых для Д11К-диагностики заболевания на развернутой стадии. При наличии возможности проведения ДНК-диагностики эта процедура становится центральным звеном медико-генетического консультирования у лиц из группы риска, трансформируя определенные теоретические величины рассчитанного риска либо в сторону многократного повышения (вплоть до 100% при обнаружении мутации), либо, напротив, в сторону полного исключения риска наследственного заболевания. Определение генотипа клинически здоровых консультируемых лиц, обозначаемое термином прогностическое (предсказательное) тестирование, представляет собой едва ли не первый пример в истории клинической медицины, демонстрирующий возможность научно обоснованного прогноза будущей судьбы клинически здорового человека за многие годы до ожидаемого дебюта тяжелого (нередко - неизлечимого) заболевания. Поэтому закономерно, что осуществление прогностического ДНК-тестирования связано с решением серьезных правовых и этических вопросов, рассматриваемых во второй части настоящей главы.
Медико-генетическое консультирование у родственников пробанда существенно осложняется возможностью новой мутации у больного со спорадическим случаем болезни. Совокупность связанных с этой возможностью проблем наиболее удобно рассмотреть на примере ПМД Дюшенна и Бекера. Как указывалось г. главе 3 (раздел 3.1), не менее трети всех случаев данных форм мышечной дистрофии обусловлены спонтанным igt; мутациями de novo в гене дистрофина [Darras В. et al 1988], в связи с чем вопросы коррекции величины генетического риска для новых мутаций являются особенно актуальными и наиболее изученными именно при ПМД Дюшенна/Бекера. В случае мутации de novo прогноз для членов консультируемой семьи с ПМД Дюшенна/Бекера зависит от того, на каком этапе гамето- или онтогенеза данная мутация возникла. Если новая мутация в гене дистрофина возникает в зрелой яйцеклетке, будет иметь место истинный спорадический случай болезни; при этом все клетки больного, имеющие одну и ту же материнскую хромосому, будут иметь идентичный мутантный генотип. Если же мутация возникает после оплодотворения (т.е. на стадии эмбриогенеза), то клетки-потомки мутантного клона будут иметь идентичную мутацию в гене дистрофина, тогда как другая популяция клеток организма останется нормальной (рис. 73). Указанное явление носит название соматического мозаиг-щзма. В случае соматического мозаицизма тяжесть и распространенность симптоматики определяются соотношением нормального и мутантного клеточных клонов организма и тем, какие ткани развиваются из мутантного клона (чем раньше в эмбриогенезе возникла мутация, тем большее число тканей будут вовлечены в патологический процесс). В обеих рассмотренных ситуациях заболевание проявляется как спорадический случай, а риск повторного появления мутантной хромосомы у братьев- ссстер пробанда не отличается от общепопуляционного.
В ряде случаев новая мутация может возникнуть в родительской половой клетке на ранней (митотической) стадии гаметогенеза - на уровне первичных половых клеток будущей матери или отца (рис 74). Это дает начало разделению клеток-потомков на нормальную и мутантную популяции гамет - феномен гонадного мо- ишцизма. Наличие гонадного мозаицизма видоизменяет расчеты риска при медико-генетическом консультировании. Так, при ПМД Дюшенна/Бекера братья и сестры больного, оцениваемого как «спорадический» случай
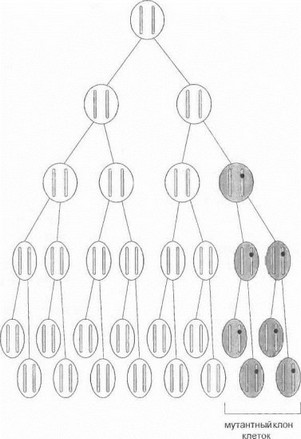
Рис. 73. Соматический мозаицизм
Мутация, возникшая de novo па одной из хромосом в соматической клетке, обозначена черным кружком.
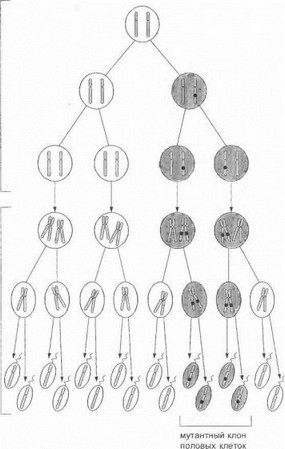
мейоз митозы клеток-предшественников
Рис. 74. Гонадный мозаицизм
Мутация, возникшая de novo на одной из хромосом в первичной половой клетке, обозначена черным кружком.
(т.е. когда у матери не выявляется гетерозиготное носи- тельство мутации), имеют генетический риск не менее 5-10% унаследовать ту же мутацию - в силу теоретической возможности того, что их мать является гонадным мозаиком [Bakker Е. et al., 1989; van Essen A. et al., 1992].
Наконец, мутация может возникнуть у будущей матери (отца) на ранней стадии их эмбрионального развития в такой клетке, которая дает начало развитию и дифференцировке зародышевых тканей. В этом случае мать (отец) будут являться соматическими мозаиками, но при этом все их гаметы будут мутантными. В такой ситуации дальнейшее наследование мутантной хромосомы у потомков (и, соответственно, наследование болезни у мужчин или наследование носительства у женщин) будет полностью подчиняться классическому X- сцепленному рецессивному типу. Родительский соматический мозаицизм, однако, является еще одним источником ошибок при медико-генетическом консультировании: поскольку обычные процедуры генодиагностики базируются на исследовании ДНК клеток крови, данный анализ может показать нормальный генотип родителя, тогда как половые клетки обследуемого родителя являются мутантными.
Наличие соматического или гонадного мозаициз- ма может быть подтверждено молекулярно-генетическими методами, например - с помощью сравнительного анализа гаплотипов больных и здоровых лиц в информативных родословных [Lebo R. et al., 1990; Melis M. et al., 1993]. В тех случаях, когда такое исследование невозможно, соответствующие поправки на мозаицизм при оценке величины генетического риска делаются на основе эмпирических данных. Например, для локуса дис- трофина в спорадических случаях ПМД Дюшенна/Бекера возможность материнского соматического/гонадного мозаицизма составляет около 7% [van Essen A. et al., 1997].
Свои особенности имеет медико-генетическое консультирование в случае новых мутаций при заболеваниях, обусловленных экспансией тринуклеотидных повторов. В главе 1 (раздел 1.3, рис. 18) подробно рассматривался механизм возникновения динамических мутаций de novo. Источником такой мутации обычно является имеющийся у здорового родителя аллель с «промежуточным» числом повторов, который в гаметогене- зе трансформируется в полную мутацию и ведет к развитию болезни у ребенка. При таком механизме генетически нестабильный «промежуточный» родительский аллель может претерпевать повторные аналогичные трансформации при передаче гена следующим детям. Таким образом, при спорадических случаях тринуклеотидных болезней, обусловленных новой мутацией у пробанда, для сибсов риск унаследовать заболевание является гораздо более высоким, чем считалось ранее [Goldberg Y. et al., 1993]. Поэтому в подобной ситуации (даже при отсутствии явной мутации у родителей!) оправданной является постановка вопроса о проведении прямой ДНК-диагностики у всех братьев-сестер пробанда. Следует помнить, что при установлении мутации de novo возможность наличия в семье наследственного заболевания при видимом отсутствии «наследственности» нередко с недоверием встречается тестйруеъ^ыми лицами, что требует от врача особого такта и аргументированности при проведении медико-генетического консультирования.
В случае выявления у плода носительства мутации (мутантной хромосомы) вопрос о продолжении или прерывании беременности должен решаться строго ин
дивидуально, с учетом всей совокупности медицинских и социальных факторов, главными из которых являются тяжесть прогнозируемого заболевания, предполагаемый возраст манифестации симптомов, возможность эффективной патогенетической терапии и т.д. При ожидаемом рождении ребенка с генотипом «высокого риска» в отношении курабельного заболевания (например, гепато- лентикулярной дегенерации) оправданной является рекомендация сохранить беременность, поскольку существующие методы превентивной терапии позволяют полностью предотвратить развитие симптомов этой тяжелой болезни (см. главу 3, раздел 3.2.3). Сложнее принять решение при выявлении у плода мутации, связанной с высоким риском развития неизлечимого заболевания. При ряде таких заболеваний (например, торсионной дистонии) пенетрантность мутантного гена не полная, а его экспрессивность может быть весьма вариабельной, в связи с чем носительство мутации само по себе отнюдь не является абсолютным предиктором манифестации у будущего ребенка тяжелых проявлений болезни. Следует помнить также, что многие заболевания (классический пример - хорея Гентингтона) проявляются обычно лишь в позднем возрасте, предоставляя носителю мутации несколько десятилетий для активной, полноценной жизни, а также оставляя реальный шанс дожить до внедрения в практику эффективных методов лечения болезни. Таким образом, в данном случае право будущей матери сохранить плод даже при получении положительного результата пренатальной ДНК-диагностики имеет под собой серьезные моральные и медицинские основания. Прерывание беременности представляется наиболее целесоообразным при однозначно высоком риске развития у будущего ребенка тяжелого некурабельного заболевания, манифестирующего в ран
нем детстве (например, при выявлении у плода мутации в гене дистрофина и прогнозируемом развитии у ребенка мышечной дистрофии Дюшенна). В любом случае окончательное решение по результатам пренатальной ДНК-диагностики всегда остается за супругами. Врач ни в коем случае не имеет права оказывать какое-либо психологическое давление; его роль заключается в детальном информировании родственников о смысле и результатах проведенных исследований, величине генетического риска, а также в объяснении (в доходчивой форме) всех нюансов существующей ситуации и прогноза.
Как уже было отмечено выше, практика проведения прогностического ДНК-тестирования у клинически здоровых лиц из группы риска привела к интенсивному развитию новой области медицинской генетики, связанной с оцешсой этических аспектов указанного вида медицинской помощи и обеспечением правовой защиты консультируемых лиц [Доклад научной группы ВОЗ, 1997]. На этапе зарождения и выработки основных принципов генетического консультирования вопросам мо- ральпо-этического порядка не всегда уделялось должное внимание. Так, считалось, что врач-генетик обязан в первую очередь выполнйРгь свой врачебный долг по отношению к обществу - проводить профилактику рождения физически неполноценных лиц в семьях, отягощенных тяжелой наследственной патологией. Первоначально во многих странах практиковался метод полного информирования лиц из групп риска о том, что их ждет в будущем, без учета психологического статуса тестируемых лиц, в категоричной форме рекомендовалось прерывание беременности и т.д. Описаны случаи дискриминации и произвола со стороны работодателей, требовавших сведения о генетическом статусе подчиненных от страховых компаний [Natowicz М. et al., 1992]. Однако по мере совершенствования общественных отношений во всех развитых странах основы концепции генетического консультирования были подвергнуты серьезному пересмо тру. Значительно большее внимание сталс уделяться соблюдению интересов отдельной семьи и каждого ее члена, права родителей иметь ребенка, права каждого человека на жизнь. Во многом это связано с достигнутым общемировым прогрессом в области соблюдения прав человека.
Непосредственным поводом к более пристальному вниманию специалистов-генетиков к вопросам этического порядка стало внедрение в практику методов, пригодных для пресимптоматичсского ДНК-тестирова- ния лиц из группы риска при таких тяжелых неизлечимых заболеваниях, как хорея Гентингтона, доминантные формы наследственных мозжечковых дегенераций, болезнь Фридрейха и т.д. В наиболее концентрированном виде весь спектр этических вопросов может быть рассмотрен на примере проведения консультирования пациентов и клинически здоровых лиц из семей, отягощенных хореей Гентингтона. В главе 3 уже отмечалось, чте данное заболевание с экспансией тринуклеотидных CAG- повторов, благодаря специфическим особенностям клиники, течения и генетики (поздний возраст начала, антиципация, эффект «отцовской передачи» и т.д.), сталс своеобразной «моделью» при разработке вопросов медико-генетического консультирования и прогностического тестирования в современной нейрогенетике, а также в решении проблем морально-этического и юридического порядка. Важнейшая из существующие проблем — возможность нанесения тяжелейшей психоэмоциональной травмы доклиническому носителю патологического гена при сообщении ему о позитивное результате ДНК-тестирования без должной психологической подготовки. Само по себе носительство гена такого заболевания как хорея Гентингтона автоматически означает, что человек обречен заболеть неизлечимым страданием (пример которого у него уже находится перед глазами в лице больного родственника), постепенно разрушающим личность, приводящим к глубокой инва- лидизации и неизбежной смерти через 15-20 лет от его начала. В литерагуре неоднократпо описаны случаи аффективного поведения и суицидальных попыток в подобных ситуациях [FarrerL., 1986; Lawson К. etal., 1996; Almqvist Е. et al., 1999; Meiser В., Dunn S., 2000]. Может ли вообще врач-генетик брать на себя функцию «вершителя человеческих судеб», выносящего в ряде случаев фактически смертный приговор клинически здоровому человеку? Сообщать ли тестируемому лицу результаты ДНК-диагностики, если он этого желает? В какой форме сообщить ему и его родственникам результаты исследования и какой процент врачебной «лжи во имя спасения» допустим в данной ситуации? Каков будет последующий трудовой анамнез лица - носителя патологического мутантного гена, особенно если работа связана с высокой степенью ответственности? Что делать, если носитель гена - несовершеннолетний? Как построить тактику дальнейшего наблюдения за таким «потенциальным больным»? Эти проблемы вызвали острейшую дискуссию не только среди генетиков, но и среди специалистов по этике и медицинской деонтологии, социологов, юристов и законодателей, психологов, непосредственно членов семей, отягощенных наследственной патологией.
Разработка организационно-методических принципов проведения медико-генетического консультирования в семьях, отягощенных хореей Гентингтона (а также любой другой тяжелой наследственной неврологической патологией), стала возможна благодаря напряженной работе специалистов различных профилей под эгидой международных профессиональных и непрофессиональных организаций по борьбе с хореей Гентингтона. В частности, весь контроль за проведением ДНК-тестирова- иия и последующего наблюдения за больными и лицамг из группы риска был возложен на непрофессиональную Всемирную Ассоциацию хореи Гентингтона ([НА), основанную в 1979 году, в которую вошли, наряду с неврологами, генетиками, психологами и специалистами по этике, больные и члены их семей. Ассоциация представляет собой объединение добровольных национальных обществ борьбы с хореей Гентингтона из б
ричной профилактики наследственной патологии (т.е. патогенетического лечения нейрогередитарных заболеваний) достаточно ограниченны, поэтому на первый план выходит предотвращение появления «генетически больного» потомства. Этой цели служит медико-генетическое консультирование - особый вид специализированной медицинской помощи, направленный на предупреждение появления повторных случаев наследственных заболеваний в отягощенных семьях.
Медико-генетическое консультирование включает несколько основных задач [Козлова С.И. и др., 1996; Бочков Н.П., 1997; Доклад научной группы ВОЗ, 1997]:
а) установление точного диагноза и типа наследования заболевания в консультируемой семье;
б) расчет генетического риска у консультируемых; родственников, включая (при возможности) точное установление их генетического статуса с помощью методов ДНК- диагностики {прогностическое тестирование);
в) определение прогноза потомства и определение наиболее эффективного способа профилактики новых случаев заболевания (в том числе с помощью пренатальной ДНК-диагностики плода на ранних сроках беременности);
г) объяснение консультируемым лицам смысла полученной и проанализированной информации, решение возникающих юридических и морально-этических проблем;
д) помощь консультируемой семье в решении целого ряда других вопросов, касающихся планирования жизни, репродуктивного поведения и возможности деторождения, психологической поддержки, социальной адаптации и т.д.
Точная диагностика наследственного заболевания нервной системы и установление типа наследования патологического фенотипа являются первым необходимым этапом медико-генетического консультирования обратившегося за помощью лица (семьи). Этот этап может осуществляться, в зависимости от конкретных медико-организационных условий, врачом общей практики, неврологом, врачом-генетиком медико-генетической консультации либо (в идеале) квалифицированным специалистом в области клинической нейрогенетики. Особенности генеалогического анализа в неврологии и постановки диагноза наследственного заболевания нервной системы подробно проанализированы в главе 1 (раздел 1.2) и в соответствующих частных разделах главы 3. Хотелось бы еще раз подчеркнуть значение детального обследования всех членов консультируемой семьи, а также необходимость выявления у родственников любых скрытых проявлений носительства мутантного гена, в том числе с использованием комплекса дополнительных лабораторно-инструментальных методов исследования.
Адекватно проведенный клинико-генеалогический анализ позволяет выделить в консультируемой семье так называемую группу риска, которая и является основным объектом мсдийо-генетического консультирования. Под группой риска понимаются родственники пробанда, которые в соответствии с типом наследования могут являться носителями мутантного гена и имеют высокий риск заболеть данным наследственным заболеванием или передать мутантный ген потомству. Например, при хорее Гентингтона, характеризующейся аутосомно-доминантным типом наследования, практически полной пенетрантностью и поздним возрастом начала болезни, к группе риска следует относить детей пробанда, а также его братьев-сестер: для каждого из этих лиц при рождении теоретический риск унаследо-
Таблица 9
Расчетные значения вероятностей фенотипов потомства при известных типах брака (для менделирующих заболеваний)
|
Тип наследо ван ия |
Генотип отца |
Генотип матери |
Вероятность фенотипов |
|||||
|
в случае рождения сыновей |
в случае рождения дочерей |
|||||||
|
больные |
здоровые носители мутации |
здоровые (мутации нет) |
больные |
здоровые носители мутации |
здоровые (мутации нет) |
|||
|
Аутосом- |
АА |
АА |
1 |
0 |
0 |
1 |
0 |
0 |
|
|
АА |
Аа |
1 |
0 |
0 |
1 |
0 |
0 |
|
доми- |
АА |
аа |
1 |
0 |
0 |
1 |
0 |
0 |
|
нантный |
Аа |
АА |
1 |
0 |
0 |
1 |
0 |
0 |
|
|
Аа |
Аа |
3/4 |
0 |
1/4 |
3/4 |
0 |
1/4 |
|
|
Аа |
аа |
1/2 |
0 |
1/2 |
1/2 |
0 |
1/2 |
|
|
аа |
АА |
1 |
0 |
0 |
1 |
0 |
0 |
|
|
аа |
Аа |
1/2 |
0 |
1/2 |
1/2 |
0 |
1/2 |
|
|
аа |
аа |
0 |
0 |
1 |
0 |
0 |
1 |
|
Аутосом- |
АА |
АА |
0 |
0 |
1 |
0 |
0 |
1 |
|
|
АА |
Аа |
0 |
1/2 |
1/2 |
0 |
1/2 |
1/2 |
|
рецесскв- |
АА |
аа |
0 |
1 |
0 |
0 |
1 |
0 |
|
ныи |
Аа |
АА |
0 |
1/2 |
1/2 |
0 |
1/2 |
1/2 |
|
|
Аа |
Аа |
1/4 |
1/2 |
1/4 |
1/4 |
1/2 |
1/4 |
|
|
Аа |
аа |
1/2 |
1/2 |
0 |
1/2 |
1/2 |
0 |
|
|
аа |
АА |
0 |
1 |
0 |
0 |
1 |
0 |
|
|
аа |
Аа |
1/2 |
1/2 |
0 |
1/2 |
1/2 |
0 |
|
|
аа |
аа |
0 |
1 |
0 |
0 |
1 |
0 |
|
X- |
А |
АА |
1 |
0 |
0 |
1 |
0 |
0 |
|
сцеп- |
А |
Аа |
1/2 |
0 |
1/2 |
1 |
0 |
0 |
|
ленный |
А |
|
|
|
|
|
|
|
|
|
|
аа |
0 |
0 |
1 |
1 |
0 |
0 |
|
доми- |
а |
АА |
1 |
0 |
0 |
1 |
0 |
0 |
|
НЭНТНЫЙ |
а |
Аа |
1/2 |
0 |
1/2 |
1/2 |
0 |
1/2 |
|
|
|
аа |
0 |
0 |
I |
0 |
0 |
1 |
|
X- |
А |
АА |
0 |
0 |
I |
0 |
0 |
1 |
|
|
А |
Аа |
1/2 |
0 |
1/2 |
0 |
1/2 |
1/2 |
|
ленный |
А |
аа |
1 |
0 |
0 |
0 |
1 |
0 |
|
рецес- |
а |
АА |
0 |
0 |
1 |
0 |
1 |
0 |
|
сивный |
а |
Аа |
1/2 |
0 |
1/2 |
1/2 |
1/2 |
0 |
|
|
а |
аа |
I |
0 |
0 |
1 |
0 |
0 |
Примечание: вероятности фенотипов в таблице представлены для случаев с
полной пенегрантностью мутантного гена А-доминантный аллель (мутантный в случае юминантных заболеваний), а — рецессивный аллель (мутантный в случае рецессивных заболеваний).
вать мутацию и заболевание составляет 50%, но при этом для здоровых лиц риск заболеть постепенно снижается с каждым прожитым десятилетием (особенно после прохождения наиболее «опасного» возраста в 40-60 лет).
При болезни Фридрейха, наследующейся по аутосом- но-рецессивному типу и начинающейся обычно на втором десятилетии жизни, основную группу риска составляют братья и сестры пробанда (теоретический риск 25%), причем степень риска заболевания выше у младших сибсов, не достигших «критического» возраста манифестации болезни. При Х-сцепленной мышечной дистрофии Дюшенна/Бекера к группе риска по заболеванию относятся лица мужского пола - родственники пробанда по материнской линии; помимо этого, сестры пробанда также имеют высокий (50%) риск унаследовать от матери мутантный ген и, оставаясь клинически здоровыми, передать заболевание мужчинам в следующее поколение.
Как видно из представленных примеров, выделение группы риска неразрывно связано с определением у данной категории лиц генетического риска - т.е. вероятности появления у них или их потомства соответствующего наследственного заболевания. Генетический риск до 5% оценивается как низкий и не считается противопоказанием к деторождению в консультируемой семье. Риск от 6 до 20% принято считать средним, а свыше 20% - высоким. При среднем и высоком риске рекомендации относительно возможности деторождения определяются в основном тяжестью медицинских и социальных последствий конкретного заболевания, а также возможностями и доступностью пренатальной ДНК-диагностики [Козлова С.И. и др., 1996].
До использования методов ДНК-диагностики генетический риск при менделирующих заболеваниях определяется путем вероятностных расчетов, основанных на закономерностях сегрегации патологического фенотипа при том или ином типе наследования. Точность расчета генетического риска зависит от информативности родословной и возможности установления генотипов консультируемых родителей. Если генотипы родителей (тип брака) известны или могут быть предположены с высокой вероятностью, оценка риска манифестации патологического фенотипа у потомков сравнительно проста - см. таблицу 9 [Козлова С.И. и др., 1996]. В необходимых случаях делается поправка на неполную пенетрантность мутантного гена, коэффициент инбридинга и некоторые другие факторы, влияющие на проявление фенотипа. Значительно сложнее расчет генетического риска при неизвестных генотипах родителей. Обычно это имеет место при наличии единичных случаев заболевания в семье, особенно если изучаемый синдром является генетически гетерогенным: в такой ситуации оценка сегрегации признаков в родословной неосуществима, и расчет генетического риска приходится проводить для различных возможных типов наследования. В этих случаях используются специальные математические формулы, оспованные на теории вероятности. Данные формулы при расчете общего риска принимают во внимание априорные факторы (менделевские закономерности предполагаемого типа наследования и сравнительную частоту возможных генетических вариантов болезни), а также фактическое наличие у консультируемого лица больных или здоровых родственников (что определяет апостериорную вероятность рассматриваемой гипотезы). При неизвестном типе брака рождение каждого последующего ребенка (здорового или больного) существенно дополняет имеющуюся генетическую информацию и видоизменяет величину апостериорной вероятности.
Нри мультифакториальных заболеваниях с поли- генно-детерминируемой наследственной предрасположенностью суммарный генетический риск зависит от большого числа факторов и определяется с помощью специальных таблиц эмпирического риска.
Возможность ДНК-диагностики существенно повышает точность определения генетического риска, позволяя от вероятностных моделей перейти к однозначному (дефинитивному) определению генотипов и ожидаемых фенотипов. В практике медико-генетического консультирования методы ДНК-анализа могут использоваться для пренатальной, преимплантационной и ранней доклинической диагностики носительства мутации. Пренатальная Д] IK-диагностика предполагает определение генотипа плода на ранних сроках беременности (пер- вый-второй триместр, предпочтительнее на 8-12-й неделе), что дает возможность при обнаружении мутации сделать медицинский аборт. Образцы ДПК плода получают из биоптатов хориона (хорионбиопсия), плаценты (плацентобиопсия), клеток амниотической жидкости (ам- ниоцентез) или лимфоцитов пуповинной крови (кордо- центез). К сожалению, все указанные процедуры, проводящиеся под контролем ультразвука, являются инвазивными и сопряжены с определенным риском прерывания беременности, оцениваемым в среднем как 1,5-2%. Новым неинвазивным методом в пренатальной диагностике является анализ ядросодержащих клеток плода, присутствующих в крови матери [Золотухина Т.В., Шилова Н.В., 1999]. Эти клетки (трофобластные клетки, лимфоциты, гранулоциты, эритробласты плода) .появляются в организме матери в результате трансплацентарного переноса, способствуя развитию толерантности материнского организма по отношению к плоду; их концентрация составляет, ориентировочно, 10-5—10 s. В настоящее время наиболее серьезной проблемой является недостаточная эффективность существующих методов сортировки и детекции плодных клеток, получаемых из крови матери. Одним из возможных решений данной проблемы является многократная амплификация генома единичных клеток плода с помощью полимеразной цепной реакции. По-видимому, перспективы более широкого использования анализа клеток плода из крови матери с целью пренатальной ДНК-диагностики связаны с дальнейшим развитием методов обогащения популяции плодных клеток.
Если в обследуемой семье мутация изучаемого гена известна (т.е. выявлена ранее у больного родственника или клинически здорового носителя), при пренатальной ДНК-диагностике плода молекулярное тестирование облегчается - например, путем использования соответствующей рестрикционной эндонуклеазы. В том случае, когда предварительный генетический материал больного члена семьи (носителя) недоступен для исследования, поиск мутаций у плода осуществляется стандартными методами мутационного скрининга - в зависимости от генетических особенностей изучаемого заболевания, подробно разбираемых в главе 3. Выявление конкретных мутаций у плода позволяет с абсолютной достоверностью устанавливать его генетический статус и определять прогноз. Несколько иначе обстоит дело с пренатальной ДНК-диагностикой, осуществляемой косвенными методами. Такая диагностика обычно проводится в тех случаях, когда изучаемый ген не идентифицирован (известна лишь его хромосомная локализация) либо он имеет сложную структуру, что технически значительно затрудняет поиск мутаций. Точность косвенной ДНК-диагностики, позволяющей оценивать наследование мутантной хромосомы, определяется близостью патологического гена и используемого маркера (маркеров). Например, если ген болезни и изучаемый маркер
расположены друг от друга на расстоянии 2 сМ, вероятность рекомбинации между данными локусами (и следовательно, вероятность ошибки) составляет 2%; соответственно, точность диагностики носительства мутантной или нормальной хромосомы в этом случае составляет около 98%. Идеальным является использование внутригенных маркеров в комбинации с тесно сцепленными маркерами, фланкирующими изучаемый ген с теломерной и центромерной сторон.
Весьма перспективным направлением профилактики наследственной патологии является преимпланта- ционная ДНК-диагностика. Сущность ее заключается в исследовании ДНК клеток раннего зародыша, развитие которого инициируется in vitro путем искусственного оплодотворения (альтернативой является анализ ДНК полярных телец овулировавших яйцеклеток). Имплантация зародыша в матку осуществляется только после исключения у него носительства мутантного гена. Таким образом исход соответствующей беременности становится, с генетической точки зрения, однозначно благоприятным. В неврологии наибольший опыт преимп- лантационной ДНК-диагностики получен при мышечной дистрофии Дюшенна/Векера, спинальной амиотрофии и некоторых других моногенных заболеваниях (Vertinsky Y., Kuliev А., 1993; Fallon L. etal., 1999]. До настоящего времени основной проблемой преимплантационной ДНК- диагностики является недостаточно высокий процент приживаемости имплантируемых в матку зародышей (не более 20-30%). Однако с развитием микрохирургической техники и общим прогрессом гинекологии и эндокринологии можно ожидать новых существенных успехов в техническом и медико-биологическом обеспечении преимплантационной диагностики наследственных заболеваний.
Методы ДНК-диагностики носительства мутаций у клинически здоровых лиц, принадлежащих к группе риска, принципиально не отличаются от аналогичных подходов, используемых для Д11К-диагностики заболевания на развернутой стадии. При наличии возможности проведения ДНК-диагностики эта процедура становится центральным звеном медико-генетического консультирования у лиц из группы риска, трансформируя определенные теоретические величины рассчитанного риска либо в сторону многократного повышения (вплоть до 100% при обнаружении мутации), либо, напротив, в сторону полного исключения риска наследственного заболевания. Определение генотипа клинически здоровых консультируемых лиц, обозначаемое термином прогностическое (предсказательное) тестирование, представляет собой едва ли не первый пример в истории клинической медицины, демонстрирующий возможность научно обоснованного прогноза будущей судьбы клинически здорового человека за многие годы до ожидаемого дебюта тяжелого (нередко - неизлечимого) заболевания. Поэтому закономерно, что осуществление прогностического ДНК-тестирования связано с решением серьезных правовых и этических вопросов, рассматриваемых во второй части настоящей главы.
Медико-генетическое консультирование у родственников пробанда существенно осложняется возможностью новой мутации у больного со спорадическим случаем болезни. Совокупность связанных с этой возможностью проблем наиболее удобно рассмотреть на примере ПМД Дюшенна и Бекера. Как указывалось г. главе 3 (раздел 3.1), не менее трети всех случаев данных форм мышечной дистрофии обусловлены спонтанным igt; мутациями de novo в гене дистрофина [Darras В. et al 1988], в связи с чем вопросы коррекции величины генетического риска для новых мутаций являются особенно актуальными и наиболее изученными именно при ПМД Дюшенна/Бекера. В случае мутации de novo прогноз для членов консультируемой семьи с ПМД Дюшенна/Бекера зависит от того, на каком этапе гамето- или онтогенеза данная мутация возникла. Если новая мутация в гене дистрофина возникает в зрелой яйцеклетке, будет иметь место истинный спорадический случай болезни; при этом все клетки больного, имеющие одну и ту же материнскую хромосому, будут иметь идентичный мутантный генотип. Если же мутация возникает после оплодотворения (т.е. на стадии эмбриогенеза), то клетки-потомки мутантного клона будут иметь идентичную мутацию в гене дистрофина, тогда как другая популяция клеток организма останется нормальной (рис. 73). Указанное явление носит название соматического мозаиг-щзма. В случае соматического мозаицизма тяжесть и распространенность симптоматики определяются соотношением нормального и мутантного клеточных клонов организма и тем, какие ткани развиваются из мутантного клона (чем раньше в эмбриогенезе возникла мутация, тем большее число тканей будут вовлечены в патологический процесс). В обеих рассмотренных ситуациях заболевание проявляется как спорадический случай, а риск повторного появления мутантной хромосомы у братьев- ссстер пробанда не отличается от общепопуляционного.
В ряде случаев новая мутация может возникнуть в родительской половой клетке на ранней (митотической) стадии гаметогенеза - на уровне первичных половых клеток будущей матери или отца (рис 74). Это дает начало разделению клеток-потомков на нормальную и мутантную популяции гамет - феномен гонадного мо- ишцизма. Наличие гонадного мозаицизма видоизменяет расчеты риска при медико-генетическом консультировании. Так, при ПМД Дюшенна/Бекера братья и сестры больного, оцениваемого как «спорадический» случай
Рис. 73. Соматический мозаицизм
Мутация, возникшая de novo па одной из хромосом в соматической клетке, обозначена черным кружком.
мейоз митозы клеток-предшественников
Рис. 74. Гонадный мозаицизм
Мутация, возникшая de novo на одной из хромосом в первичной половой клетке, обозначена черным кружком.
(т.е. когда у матери не выявляется гетерозиготное носи- тельство мутации), имеют генетический риск не менее 5-10% унаследовать ту же мутацию - в силу теоретической возможности того, что их мать является гонадным мозаиком [Bakker Е. et al., 1989; van Essen A. et al., 1992].
Наконец, мутация может возникнуть у будущей матери (отца) на ранней стадии их эмбрионального развития в такой клетке, которая дает начало развитию и дифференцировке зародышевых тканей. В этом случае мать (отец) будут являться соматическими мозаиками, но при этом все их гаметы будут мутантными. В такой ситуации дальнейшее наследование мутантной хромосомы у потомков (и, соответственно, наследование болезни у мужчин или наследование носительства у женщин) будет полностью подчиняться классическому X- сцепленному рецессивному типу. Родительский соматический мозаицизм, однако, является еще одним источником ошибок при медико-генетическом консультировании: поскольку обычные процедуры генодиагностики базируются на исследовании ДНК клеток крови, данный анализ может показать нормальный генотип родителя, тогда как половые клетки обследуемого родителя являются мутантными.
Наличие соматического или гонадного мозаициз- ма может быть подтверждено молекулярно-генетическими методами, например - с помощью сравнительного анализа гаплотипов больных и здоровых лиц в информативных родословных [Lebo R. et al., 1990; Melis M. et al., 1993]. В тех случаях, когда такое исследование невозможно, соответствующие поправки на мозаицизм при оценке величины генетического риска делаются на основе эмпирических данных. Например, для локуса дис- трофина в спорадических случаях ПМД Дюшенна/Бекера возможность материнского соматического/гонадного мозаицизма составляет около 7% [van Essen A. et al., 1997].
Свои особенности имеет медико-генетическое консультирование в случае новых мутаций при заболеваниях, обусловленных экспансией тринуклеотидных повторов. В главе 1 (раздел 1.3, рис. 18) подробно рассматривался механизм возникновения динамических мутаций de novo. Источником такой мутации обычно является имеющийся у здорового родителя аллель с «промежуточным» числом повторов, который в гаметогене- зе трансформируется в полную мутацию и ведет к развитию болезни у ребенка. При таком механизме генетически нестабильный «промежуточный» родительский аллель может претерпевать повторные аналогичные трансформации при передаче гена следующим детям. Таким образом, при спорадических случаях тринуклеотидных болезней, обусловленных новой мутацией у пробанда, для сибсов риск унаследовать заболевание является гораздо более высоким, чем считалось ранее [Goldberg Y. et al., 1993]. Поэтому в подобной ситуации (даже при отсутствии явной мутации у родителей!) оправданной является постановка вопроса о проведении прямой ДНК-диагностики у всех братьев-сестер пробанда. Следует помнить, что при установлении мутации de novo возможность наличия в семье наследственного заболевания при видимом отсутствии «наследственности» нередко с недоверием встречается тестйруеъ^ыми лицами, что требует от врача особого такта и аргументированности при проведении медико-генетического консультирования.
В случае выявления у плода носительства мутации (мутантной хромосомы) вопрос о продолжении или прерывании беременности должен решаться строго ин
дивидуально, с учетом всей совокупности медицинских и социальных факторов, главными из которых являются тяжесть прогнозируемого заболевания, предполагаемый возраст манифестации симптомов, возможность эффективной патогенетической терапии и т.д. При ожидаемом рождении ребенка с генотипом «высокого риска» в отношении курабельного заболевания (например, гепато- лентикулярной дегенерации) оправданной является рекомендация сохранить беременность, поскольку существующие методы превентивной терапии позволяют полностью предотвратить развитие симптомов этой тяжелой болезни (см. главу 3, раздел 3.2.3). Сложнее принять решение при выявлении у плода мутации, связанной с высоким риском развития неизлечимого заболевания. При ряде таких заболеваний (например, торсионной дистонии) пенетрантность мутантного гена не полная, а его экспрессивность может быть весьма вариабельной, в связи с чем носительство мутации само по себе отнюдь не является абсолютным предиктором манифестации у будущего ребенка тяжелых проявлений болезни. Следует помнить также, что многие заболевания (классический пример - хорея Гентингтона) проявляются обычно лишь в позднем возрасте, предоставляя носителю мутации несколько десятилетий для активной, полноценной жизни, а также оставляя реальный шанс дожить до внедрения в практику эффективных методов лечения болезни. Таким образом, в данном случае право будущей матери сохранить плод даже при получении положительного результата пренатальной ДНК-диагностики имеет под собой серьезные моральные и медицинские основания. Прерывание беременности представляется наиболее целесоообразным при однозначно высоком риске развития у будущего ребенка тяжелого некурабельного заболевания, манифестирующего в ран
нем детстве (например, при выявлении у плода мутации в гене дистрофина и прогнозируемом развитии у ребенка мышечной дистрофии Дюшенна). В любом случае окончательное решение по результатам пренатальной ДНК-диагностики всегда остается за супругами. Врач ни в коем случае не имеет права оказывать какое-либо психологическое давление; его роль заключается в детальном информировании родственников о смысле и результатах проведенных исследований, величине генетического риска, а также в объяснении (в доходчивой форме) всех нюансов существующей ситуации и прогноза.
Как уже было отмечено выше, практика проведения прогностического ДНК-тестирования у клинически здоровых лиц из группы риска привела к интенсивному развитию новой области медицинской генетики, связанной с оцешсой этических аспектов указанного вида медицинской помощи и обеспечением правовой защиты консультируемых лиц [Доклад научной группы ВОЗ, 1997]. На этапе зарождения и выработки основных принципов генетического консультирования вопросам мо- ральпо-этического порядка не всегда уделялось должное внимание. Так, считалось, что врач-генетик обязан в первую очередь выполнйРгь свой врачебный долг по отношению к обществу - проводить профилактику рождения физически неполноценных лиц в семьях, отягощенных тяжелой наследственной патологией. Первоначально во многих странах практиковался метод полного информирования лиц из групп риска о том, что их ждет в будущем, без учета психологического статуса тестируемых лиц, в категоричной форме рекомендовалось прерывание беременности и т.д. Описаны случаи дискриминации и произвола со стороны работодателей, требовавших сведения о генетическом статусе подчиненных от страховых компаний [Natowicz М. et al., 1992]. Однако по мере совершенствования общественных отношений во всех развитых странах основы концепции генетического консультирования были подвергнуты серьезному пересмо тру. Значительно большее внимание сталс уделяться соблюдению интересов отдельной семьи и каждого ее члена, права родителей иметь ребенка, права каждого человека на жизнь. Во многом это связано с достигнутым общемировым прогрессом в области соблюдения прав человека.
Непосредственным поводом к более пристальному вниманию специалистов-генетиков к вопросам этического порядка стало внедрение в практику методов, пригодных для пресимптоматичсского ДНК-тестирова- ния лиц из группы риска при таких тяжелых неизлечимых заболеваниях, как хорея Гентингтона, доминантные формы наследственных мозжечковых дегенераций, болезнь Фридрейха и т.д. В наиболее концентрированном виде весь спектр этических вопросов может быть рассмотрен на примере проведения консультирования пациентов и клинически здоровых лиц из семей, отягощенных хореей Гентингтона. В главе 3 уже отмечалось, чте данное заболевание с экспансией тринуклеотидных CAG- повторов, благодаря специфическим особенностям клиники, течения и генетики (поздний возраст начала, антиципация, эффект «отцовской передачи» и т.д.), сталс своеобразной «моделью» при разработке вопросов медико-генетического консультирования и прогностического тестирования в современной нейрогенетике, а также в решении проблем морально-этического и юридического порядка. Важнейшая из существующие проблем — возможность нанесения тяжелейшей психоэмоциональной травмы доклиническому носителю патологического гена при сообщении ему о позитивное результате ДНК-тестирования без должной психологической подготовки. Само по себе носительство гена такого заболевания как хорея Гентингтона автоматически означает, что человек обречен заболеть неизлечимым страданием (пример которого у него уже находится перед глазами в лице больного родственника), постепенно разрушающим личность, приводящим к глубокой инва- лидизации и неизбежной смерти через 15-20 лет от его начала. В литерагуре неоднократпо описаны случаи аффективного поведения и суицидальных попыток в подобных ситуациях [FarrerL., 1986; Lawson К. etal., 1996; Almqvist Е. et al., 1999; Meiser В., Dunn S., 2000]. Может ли вообще врач-генетик брать на себя функцию «вершителя человеческих судеб», выносящего в ряде случаев фактически смертный приговор клинически здоровому человеку? Сообщать ли тестируемому лицу результаты ДНК-диагностики, если он этого желает? В какой форме сообщить ему и его родственникам результаты исследования и какой процент врачебной «лжи во имя спасения» допустим в данной ситуации? Каков будет последующий трудовой анамнез лица - носителя патологического мутантного гена, особенно если работа связана с высокой степенью ответственности? Что делать, если носитель гена - несовершеннолетний? Как построить тактику дальнейшего наблюдения за таким «потенциальным больным»? Эти проблемы вызвали острейшую дискуссию не только среди генетиков, но и среди специалистов по этике и медицинской деонтологии, социологов, юристов и законодателей, психологов, непосредственно членов семей, отягощенных наследственной патологией.
Разработка организационно-методических принципов проведения медико-генетического консультирования в семьях, отягощенных хореей Гентингтона (а также любой другой тяжелой наследственной неврологической патологией), стала возможна благодаря напряженной работе специалистов различных профилей под эгидой международных профессиональных и непрофессиональных организаций по борьбе с хореей Гентингтона. В частности, весь контроль за проведением ДНК-тестирова- иия и последующего наблюдения за больными и лицамг из группы риска был возложен на непрофессиональную Всемирную Ассоциацию хореи Гентингтона ([НА), основанную в 1979 году, в которую вошли, наряду с неврологами, генетиками, психологами и специалистами по этике, больные и члены их семей. Ассоциация представляет собой объединение добровольных национальных обществ борьбы с хореей Гентингтона из б