
В силу особенностей генетики митохондриальных энцефаломиопатий ДНК-диагностика этих заболеваний имеет ряд принципиальных отличий от традиционных подходов, применимых для менделирующих заболеваний. Обобщение многолетнего опыта обследования больных с митохондриальной патологией в ведущих исследовательских центрах мира позволило разработать четкий и последовательный диагностический алгоритм, обеспечивающий наибольшую эффективность диагностического поиска [Краснопольская К.Д., Захарова Е.Ю., 1998; Shoffner .!., Wallace D., 1992; DiMauro S., 1993; Chinnery P. et al., 1999 (а)]. Данный алгоритм включает несколько основных этапов (рис. 69).
- На первом этапе проводится детальный клинико-генеалогический и лабораторно-инструментальный анализ, целью которого является накопление доказательств в пользу обоснованного предположения о митохондриальной природе изучаемого заболевания. Наиболее очевидно о митохондриальной болезни может свидетельствовать: а) материнский тип наследования (с учетом всех полиморфных и даже субклинических проявлений у сибсов - детей больной матери); б) своеобразный характер синдрома (наличие мультисистемной и мультиорганной патологии с вовлечением органов, различных по эмбриональному происхождению и функциям); в) прогрессирующее течение, наличие метаболических кризов (последнее особенно актуально для митохондриальных синдромов раннего детского возраста); г) повышение уровня лактата в крови и спинномозговой жидкости (в том числе на фоне пищевой и физической нагрузки); д) аминоацидурия и органическая ацидурия. Поиск мультиорганной патологии должен проводиться целенаправленно, с использованием необходимых параклинических методов, с целью выявления явной или скрытой кардиомиопатии, почечной тубулопатии, гепа- тоцеллюлярной дисфункции, диабета, недостаточности гормона роста, атрофии ворсин кишечника, изменений в мазке крови и пунктате костного мозга, миопатии, периферической, кохлеарной и зрительной невропатии, патологии сетчатки, петрификатов и очаговых изменений в веществе мозга.
- Уже в результате проведенного вышеуказанного комплексного обследования может быть достаточно четко идентифицирован один из известных клинических синдромов, обычно обусловленных толковыми мутациями мтДНК (MELAS, MERRF, NARP, атрофия зрительных нервов Лебера). В таком случае ДНК из клеток крови может исследоваться на наличие соответствующих известных мутаций мтДНК. Молекулярный анализ облегчается тем, что каждый из указанных синдромов ассоциирован со сравнительно ограниченным числом мутаций мтДНК, часть которых являются мажорными и имеются у абсолютного большинства больных. 11ри выявлении искомой мутации в лимфоцитах диагноз может считаться окончательно подтвержденным.

Однако такой наиболее благоприятный «сценарий» на практике реализуется далеко не всегда.
- На рассматриваемых этапах основная проблема заключается в том, что легко доступные клетки крови не являются идеальным источником для поиска мутаций мтДНК. Клетки кроветворной системы размножаются весьма быстро, поэтому в силу митотической сегрегации мутантной мгДНК ее содержание в крови весьма вариабельно (от 0% до 60-80%). Более того, в эмбриогенезе кроветворный росток может вообще не унаследовать мтДНК мутантного вида, тогда как при этом в пораженных тканях (мозг, мышцы и др.) число копий мутантной мтДНК будет патогенетически значимым. Например, пациенты с лебсровской атрофией зрительных нервов и MERRF обычно имеют достаточно высокое содержание мутантной мтДНКв лимфоцитах (что облегчает ДНК-диагностику этих болезней), тогда как при MELAS и особенно синдроме Кернса-Сейра ситуация обратная. Таким образом, отсутствие мутаций при исследовании ДНК лимфоцитов отнюдь не исключает митохондриальной природы болезни. Гораздо более информативным источником являются скелетные мышцы, поскольку, во-первых, отсутствие клеточных деленйй в данной постмитотической ткани способствует «удержанию» митохондрий с мутантной мтДНК, а, во-вторых, мышечная ткань является одной из основных «мишеней» патологического процесса при митохондриальных энцефаломиопатиях. Поэтому следующий шаг при обследовании больного с неидентифицированной мутацией предполагает проведение биопсии скелетной мышцы (обычно четырехглавой или дельтовидной).
- Образцы мышечных биоптатов целесообразно делить на 3 части - одна для микроскопического исследования (гистология, гистохимия и электронная микро- скопил), вторая для энзимологического и иммунологического анализа (изучение характеристик компонентов дыхательной цепи) и третья - для молекулярно-генетического анализа. Как и при исследовании ДНК крови, анализ мтДНК мышцы начинается с исследования известных толковых мутаций или перестроек, специфических для изучаемого синдрома. Поиск известных мутаций на мышечном материале позволяет в большинстве случаев успешно осуществлять ДНК-диагностику болезни. Следует добавить, что обычно наряду с мутантной мтДНК в мышце выявляется нормальный вид мтДНК, и в такой ситуации весьма ценным является дополнительное определение уровня гетероплазмии (анализ дозы мутантного гена с помощью количественной ПЦР или блот-гибридизации по Саузерну). Установление процентного соотношения между нормальной и мутантной мтДНК позволяет на молекулярном уровне объективизировать тяжесть поражения и оценить прогноз заболевания.
- При отсутствии известных мутаций мтДНК в мышечной ткани следующим этапом молекулярно-генетического анализа является секвенирование всей цепи мтДНК. Это исследование, однако, является не только достаточно трудоемким и дорогостоящим, но и связано с определенными сложностями в интерпретации получаемых результатов. Поэтому предварительно необходимо провести весь комплекс морфогистохимических и биохимических анализов мышечного би оплата с целью получения абсолютно достоверных данных, позволяющих подтвердить первичную митохондриальную патологию и по возможности установить конкретное звено митохондраильной дисфункции. Так например, важными маркерами могут служить выявляемые цитохром-с- ОкСТщавно-негативные и «рваные красные» волокна, нарушение окисления пирувата и скорости синтеза АТФ, дефекты активности отдельных субъединиц комплекса дыхательной цепи и т.д. В ряде случаев указанные нарушения позволяют весьма точно локализовать биохимический уровень поражения и предположительно установить ген или группу генов мтДНК, в которых может иметь место мутация.
- Наконец, проводится секвенирование митохондриального генома с целью выявления нового варианта мутации мтДНК. Как уже указывалось, особенностью мтДНК является высокая частота спонтанных мутационных событий, поэтому большую сложность представляет обоснование патогенетической значимости той или иной новой нуклеотидной вариации, выявленной у обследуемого больного. Основными доказательствами того, что обнаруженная и неизвестная ранее му гация мтДНК является причиной болезни, считаются следующие признаки: а) гетероплазмия по данной мутации (гетероплаз- мия свидетельствует о сравнительно недавнем происхождении мутации либо о том, что она несовместима с жизнью в состоянии гомоплазмии); б) более высокое содержание мутантной мтДНК в пораженных тканях по сравнению с интактными; в) локализация мутации в эволю- ционно консервативной области мтДНК, особенно если соответствующая аминокислотная замена может существенно наруши ть структуру и функцию генного продукта; г) отсутствие данной мутации в нормальной популяции (группе контроля). Последние 2 критерия применимы и для мутаций, выявляемых в гомоплазмическом состоянии [Chinnery Р. el el., 1999 (б)].
С технической точки зрения, скрининг на известные мутации проводится на основе метода ПЦР: амп- лифицированный изучаемый участок мтДНК подвергается обработке подходящей рестрикционной эндонукле
азой либо гибридизации со специфическим олигонуклеотидом. Пример такой ДНК-диагностики при синдроме MELAS на примере мажорной мутации A3243G показан на рис. 70.
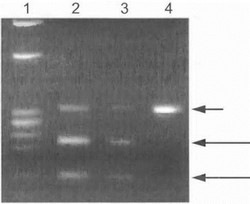
Рисунок70. Прямая ДНК-диагностика мутации 3243А—gt;G при синдроме MELAS
Исследование проведено на г еномной ДНК, продукты амплификации обработаны рсстриктазой/1/я/1. Дорожка 1 - маркер, дорожки 2иЗ-больные с синдромом MELAS, имеющие мутацию 3243А—gt;Й вгетероплазмическом сос тоянии (данная мутация создает сайт рестрикции для фермента Ара I), дорожка 4 здоровый контроль. Нормалы пай фрагмент сшплифшц ipoBai той МТД Н К имеет длину 330 п.о. (короткая стрелка), мутантная мтДНК разрезается ферментом на 2 фрагмента длиной 213 и 117 п.о. (длинные стрелки).
Перестройки и количественные дефекты мтДНК (единичные и множественные делеции, дупликации, истощение мтДНК) регистрируются методом блот-гибри- дизации по Саузерну после обработки мтДНК рестрикционными эндонуклеазами (рис. 68 A-В) В последние годы в практику все шире входят новые высокоскоростные методы анализа ДНК, значительно облегчающие молекулярную диагностику (автоматическое секвенирова- ние, использование биологических микрочипов и др.).
Идентификация генетического дефекта у пробанда дает возможность проведения ДНК-диагностики у родственников в группе риска - в первую очередь у бра- тьев-сестер пробанда и у детей больной матери. В принципе, при митохондриальном наследовании все сибсы - дети одной матери являются носителями определенного количества мутантной мтДНК, однако ее содержание в крови и других тканях у сибсов чрезвычайно вариабельно. Поэтому анализ уровня гетероплазмии позволяет с известной осторожностью оценивать относительный риск, прогноз возможного заболевания и необходимость превентивного лечения у клинически здоровых лиц. Что касается пренатальной ДНК-диагностики, то ее проведение в традиционном виде при митохондриальных болезнях в настоящее время не рекомендуется. Дело в том, что содержание мутантной мтДНК в ворсинах хориона не дает однозначного ответа i1*l вопрос о характере поражения мозга и других тканей плода, а также о дальнейшей динамике митотической сегрегации мутантной мтДНК в тканях на протяжении жизни [Poulton J. et al., 1998]. В литератере описаны отдельные случаи искусственного прерывания беременности при выявлении в ворсинах хориона крайне высокого содержания исследуемой митохондриальной мутации [Harding A. et al., 1992], однако этот вопрос до последнего времени остается дискуссионным. Альтернативой может быть преимпланта- ционная ДНК-диагностика, позволяющая в некоторых случаях накануне фертилизации in vitro проводить предварительный отбор яйцеклеток, не содержащих определяемого количества мутантной мтДНК [Poulton J. etal., 19981.
Наконец, поистине революционизирующая статья была опубликована в 1997 году в журнале «Lancet»: авторы сообщили об успешном рождении здорового ребенка после процедуры замещения цитоплазмы яйцеклетки, содержащей мутантные митохондрии, цитоплазмой донорской яйцеклетки и последующего оплодотворения in vitro [Cohen J. et ah, 1997]. Этот метод, однако, представляющий собой, по существу, «митохондриальное клонирование», связан с серьезными и вряд ли разрешимыми в обозримом будущем этическими проблемами.
Диагностика мутаций в ядериых генах при митохондриальных энцефаломиопатлях, наследующихся по менделевскому типу, широкого распространения не получила и используется лишь для решения специальных исследовательских задач в небольшом числе профильных лабораторий На практике более доступным и целесообразным методом специфической диагностики данной подгруппы митохондриальных болезней является соответствующий биохимический скрининг.