
Большинство митохондриальных белков кодируются ядерным геномом, поэтому известно значительное число митохондриальных болезней, наследуемых в соответствии с менделевскими типами наследования [Вель- тищев Ю.Е., Темин П.А., 1998; Краснопольская К.Д., Захарова Е.Ю., 1998; DiMauro S., 1993]. Биохимическая классификация данных болезней предполагает их подразделение на следующие классы:
- Дефекты транспорта митохондриальных субстратов. К ним относятся синдромы дефицита кар- нитина и ферментов его метаболизма (карнитин-паль- митоилтрансферазы, карнитин-ацилкарнитин-транслока- зы и др.). Примером заболевания с идентифицированным генетическим дефектом является аутосомно-рецес- сивный синдром недостаточности карнитин-пальмито- илтрансферазы, обусловленный мутациями гена данного фермента на хромосоме 1р.
- Дефекты утилизации митохондриальных субстратов. К ним относятся дефекты ферментов р-окисле- ния жирных кислот (соответствующие мутантные гены локализованы на хромосомах 1,2 и 12), недостаточность пируват-карбоксилазы, а также дефекты компонентов пируват-дегидрогеназного комплекса - пируват-декар- боксилазы (гены субъединиц аир на хромосомах X и
- , дигидролипоил-трансацетилазы и дигидролипоил- дегидрогеназы (один из генов на хромосоме 7). Все формы наследуются па аутосомно-рецессивному либо X- сцепленному рецессивному типам.
- Дефекты ферментов цикла Кребса - фумара- зы, сукцинат-дегидрогеназы и а-кетоглутарат-дегидрогеназы (гены локализованы на хромосомах 1 и 7). Тип наследования ферментопатий цикла Кребса аутосомно- рецессивный.
- Дефект сопряжения окисления и фосфорилирования (болезнь Люфта). Этот редчайший синдром описан лишь у 2 неродственных больных и обусловлен неспособностью митохондрий конвертировать энергию окисляемых органических субстратов в АТФ, что сопровождается гипертермией, непереносимостью тепла, профузным потом, нолифагией, полидипсией, тахикардией. Тип наследования и мутантный ген не установлены.
- Дефекты дыхательной цепи. Комплексы I-V дыхательной цепи митохондрий содержат приблизительно 83 белковых субъединицы, причем 69 из них кодируются ядерным геномом. В связи с этим известно довольно большое число клинических синдромов недостаточности комплексов I-V, наследуемых по менделевскому типу и предположительно обусловленных мутациями
] енов ядерной ДНК. Однако на сегодняшний день в литературе описано лишь несколько случаев выявления мутаций в ядерных генах, кодирующих белки дыхательной цепи и локализованных, в частности, на хромосомах 3, 5, 9, 11 и 22. Такие мутации идентифицированы в генах AQDQ, NDUFS8 и NDUFV1 комплекса I, сукци- натдегидрогеназы комплекса II, а также SURF1 и SC02 вспомогательных белков, участвующих в биогенезе комплекса IV (сборка субъединиц комплекса, поддержание его пространственной организации и т.п.).
Клиническая картина митохондриальных болезней, обусловленных мутациями ядерных генов, чрезвычайно вариабельна. Первые симптомы появляются чаще всего в раннем детском возрасте, реже болезнь манифестирует в более позднем периоде. Характерны следующие основные фенотипы: а) подострая некротизирующая энцефалопатия (болезнь Лея); б) различные варианты фатальной детской мультисистемпой энцефаломи- опатии с лактат-ацидозом, нарушениями дыхания и нередким поражением сердечной мышцы; в) прогрессирующая миоклоническая эпилепсия (судороги обычно резистентны к антиконвульсантной терапии); г) синдром Кериса-Сейра; д) прогрессирующая детская полиодист- рофия (синдром Альперса) - комбинированная дегенерация серого вещества головного мозга; е) прогрессирующая лейкодистрофия; ж) доброкачественная детская митохондриальная миопатия, непереносимость физической нагрузки; з) синдром миалгии-миоглобинурии [Вель- гищев Ю.Е., Темин П.А., 1998; Краснопольская К.Д., Захарова E.IO., 1998; DiMauro S., 1993; Chiimery Р. etal., 1999 (а)]. При многих формах неврологическая патология сопровождается поражением сердца (кардиомиопатия), печени (печеночная недостаточность, гепатомега- .пия, цирроз), почек (проксимальная тубулопатия), кишечника (синдром мальабсорбции, потеря массы тела), метаболическими кризами (гипогликемические и аци- дотические приступы с рвотой, диареей, расстройствами сознания), разнообразными аномалиями развития.
Особую группу составляют заболевания, при которых можно предполагать нарушение взаимодействий между ядерным и митохондриальным геномами. Одно из них - синдром множественных делеций мтДНК. Тип наследования чаще всего аутосомно-доминантный, в типичных случаях симптомы дебютируют на 2-3-м десятилетии жизни. В клинической картине наблюдаются весьма полиморфные мультисистемкые проявления: прогрессирующая наружная офтальмоплегия, миопатичес- кий синдром с вовлечением дыхательной мускулатуры, атаксия, нистагм, невропатии периферических, слуховых и зрительных нервов, катаракта, гипопаратиреоз, задержка роста, липоматоз и др. На компьютерных томограммах выявляются очаги пониженной плотности в базальных ганглиях, семиовальном центре и ножках мозга. При лабораторных исследованиях выявляется лактаг-ацидоз в крови, феномен «рваных красных волокон» и снижение активности цитохром с-оксидазы в мышечных био- птатах. Прогноз неблагоприятный, больные обычно погибают в течение 15-20 лет вследствие дыхательных нарушений. Кардинальным признаком данного заболевания являются сочетанные делении многих участков митохондриального генома, грубо нарушающие структуру мтДНК и функционирование различных митохондриальных генов (рис. 68Б) [Zeviani М. et al., 1989]. Предполагается, что в основе такого типа мутаций лежит повреждение регуляторных белков, кодируемых ядерными генами и контролирующих митохондриальный биогенез, в частности-процессы репликации мтДНК [DiMauro S., 1993; Carrozzо R. et al., 1998]. При исследовании больших семей с аутосомно-доминантным синдромом множественных делеций мтДНК была показана его гетерогенность: три генетических локуса данного синдрома картированы на хромосомах 10q24(PEOl), Зр14.1—21.2 (РЕ02) и 4q35 (РЕОЗ) [Chinnery Р. et al., 1999 (а); Kaukonen J. et al., 2000]. Ген последней формы идентифицирован: он кодирует фермент аденинового обмена аденин-нуклеотид-транслоказу 1 (ANT1); нарушение функции данного фермента в результате мутаций ANT1 сопровождается патологией метаболизма аденинового нуклеотида и нарушением репликации мтДНК [Kaukonen J. et al., 2000].
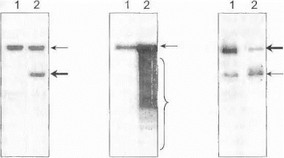
А Б В
Рис. 68. Диагностика перестроек и количественных дефектов мтДНК а помощью блот-гибридизации по Саузерну Дорожка I - здоровый контроль, дорожка 2- больной. А. Деления мтДНК у больного с синдромом Кернса -Сэпра. Тонкой с трелкой обозначена нормальная мтДНК, жирной стрелкой—мутан тная укороченная молекула мтДНК с Делением 4977 п.о. Б. Синдром множественных Делеций мтДНК. Ст релкой обозначена нормальная молекула мтДНК, скобкой - мутантные укороченные молекулы с делениями различной длины. В. Синдром истощения мтДНК Тонкой стрелкой обозначены фрагменты ядериой ДНК, служащие внутренним стандартом; жирной стрелкой обозначена мтДНК: у больного ((дорожка 2) по сравнению со здоровым индивидом четко определяется заметно сниженная интенсивность сигнала, свидетельствующая о количественном дефекте мтДНК.
Известна аутосомно-рецессивная форма синдрома множественных делеций мтДНК - так называемая митохондриальная нейрогастроинтестинальная энцефа- ломиопатия (MNGIE). Заболевание манифестирует в более молодом возрасте и проявляется тем же набором типичных для митохондриальных болезней неврологических симптомов в сочетании с выраженной дисфункцией желудочно-кишечного тракта (синдром кишечной нсевдообструкции с приступами повторной рвоты, диареей.. потерей массы тела). У больных MNGIE выявлено выраженное снижение активности фермента тимидин- фосфорилазы, обусловленное мутациями соотвегст вую- щего гена на хромосоме 22ql3.32-qter [Nishmo I. et al., 1999]. Таким образом, в основе болезни лежит генетически детерминированная патология метаболизма тими- дина, приводящая к нарушению репликации и/или поддержания молекулы мтДНК.
Синдром истощения мтДНК представляет собой состояние, пои котором у больных имеется не качественный, а количественный дефект мтДНК - т.е. резкое снижение числа копий молекул мтДНК (рис. 68 В) [Moracs С. et al., 1991; DiMauro S., 1993]. Характерно, что истощение мтДНК наблюдается только в строго определенных тканях (например, только в мышцах, только в печени, в мышцах и почках и т.д.). Клиническая картина зависит от вовлечения конкретных тканей и обычно включает в различных комбинациях миопатию (в том числе врожденную), судорожный синдром, печеночную и почечную недостаточноть, кардиомиопатию. Характерен лактат- ацидоз, феномен «рваных красных волокон», обнаруживается комбинированная недостаточность комплексов дыхательной цепи, содержащих мтДНК-кодируемые субъединицы (I, III-V). Заболевание носит врожденный характер или манифестирует на 1-2-м году жизни и имеет обычно фатальное течение. В большинстве описанных случаев синдрома истощения мтДНК зарегистрирован аутосомно-рецессивный тип наследования. Генетический дефект не установлен. Предполагается, что заболевание обусловлено повреждением ядерного гена, контролирующего репликацию мтДНК. Известна также относительно доброкачественная вторичная форма данного синдрома, при которой истощение мтДНК в клетках вызвано применением анти-ВИЧ препарата зидовудина.