
Нервно-мышечные болезни занимают первое место по частоте среди всех наследственных моногенных неврологических заболеваний. По данным A. Emery (1991), суммарная распространенность всех наследственных нервно-мышечных болезней составляет 250-300 х 10_6 (т.е. приблизительно 1 случай на 3500 населения) Принимая во внимание тот факт, что для основной части заболеваний из данной группы характерно неуклонно-прогрессирующее течение и отсутствие эффективных методов лечения, нервно-мышечные болезни следует признать одной из наиболее актуальных проблем клинической неврологии. Профилакт ика повторных случаев нервно-мышечных болезней в семьях «высокого риска» является на сегодняшний день единственным эффек ¦ тивным средством борьбы с этими тяжелыми и нередко фатальными недугами, при этом центральное место в системе профилактических мероприятий занимает ДНК диагностика.
- Прогрессирующие мышечные дистрофии
Прогрессирующие мышечные дистрофии (ПМ/ \ gt; представляют собой клинически и генетически гетеро
генную группу наследственных заболеваний, характеризующихся первичным поражением скелетной мускулатуры невоспалительного характера. Распространенность данных заболеваний весьма высока - около 200 случаев па 1 млн. населения [Emery А., 1991]. Общей характеристикой ПМД является развитие нарастающих мышечных атрофий и парезов вследствие прогрессирующей дегенерации миоцитов, обусловленной поражением структурных белков сарколеммы или ключевых ферментов скелетных мышц Прогресс в изучении данной группы заболеваний связан с раскцытием в последние годы структурных и молекулярных основ, регулирующих функционирование мышечного волокна [Иллариошкин С.Н., Иванова-Смоленская И.А., 1998; Campbell К., 1995; Worton R , 1995; Emery А., 1998].
В настоящее время клиническая классификация 11МД базируется, главным образом, на характере распределения мышечных атрофий и парезов (копечпостно- поясные, дистальные, лице-лопаточно-плечевые, оку- нофарингеальные ПМД), который дополняется типом наследования болезни (аутосомные и Х-сцепленные формы). Более детальная классификация в рамках данных I рупп ПМД предполагает идентификацию первичного молекулярного дефекта, лежащего в основе болезни.
1.1.1. Х-сцепленные ПМД Дюшенна и Бекера (дистрофинопатии)
Прогрессирующие мышечные дистрофии Дюшен- ini и Бекера относятся к самым частым формам мышеч- них дистрофий (соответственно, 1 на 3500 и 1 на 20 000 Iчищенных мальчиков) и наследуются по Х-сцепленно- му рецессивному типу [Emery А., 1991]. Форма Дюшен- п(1 начинается в возрасте 3-6 лет со слабости мышц та- юного пояса и проксимальных отделов ног, нередко сопровождающейся псевдогипертрофиями икроножных, ягодичных, дельтовидных и других мышц, кардиомиопатией. В дальнейшем характерна быстрая генерализация парезов и атрофий, нарастающая обездвиженность, развитие контрактур и дыхательных нарушений; гибель больных наступает обычно на 2-3-м десятилетии жизни [Темин П.А. и др., 1997; Emery А., 1993; Specht L., Kunkel L., 1993]. Форма Бекера традиционно рассматривается как «мягкий» клинический вариант ПМД Дю- шенна с более поздним началом болезни (в 12-15 лет), относительно доброкачественным течением и сохранностью способности к самостоятельной ходьбе на протяжении 15-20 лет от момента появления первых симптомов [Темин П.А. и соавт., 1997]. Женщины, являющиеся гетерозиготными носительницами мутантного гена, как правило, остаются клийически здоровыми, однако иногда у них могут наблюдаться отдельные субклинические проявления носительства мутации - умеренно выраженная мышечная слабость, увеличение объема икроножных мышц, высокий уровень в крови мышечного фермента креатинфосфокиназы [Emery А., 1993; Specht L., Kunkel L., 1993].
) ТМД Дюшенна и Бекера являются аллельными заболеваниями и обусловлены мутациями одного гена в хромосомном локусе Хр21 [Goodfellow Р. et ah, 1985; Monaco A. et ah, 1986; Koenig M. et ah, 1987]. Данный ген является самым большим из известных на сегодня генов человека и имеет весьма сложную молекулярную организацию: он содержит, как минимум, 5 промоторов, свыше 80 экзонов, состоит из 24 000 кб и кодирует белок с молекулярной массой 427 килодальтон, получивший название «дистрофии» [Koenig М. et ah, 1987; Hoffman Е. et ah, 1992]. В норме в мышечном волокне дистрофии локализуется на цитоплазматической поверхности сарколеммы, являясь важной составной частью цитоскелета и обеспечивая связь между актиновыми филаментами (т.е. сократительным аппаратом мышечного волокна) и сарколеммой [Ahn А., KunkelL., 1993; Campbell К., 1995; Hoffman Е., Kunkel L., 1989]. Известна также изоформа дистрофина, экспрессирующаяся в центральной нервной системе [Tinsley J. et al., 1994]; с отсутствием данной изоформы белка в мозге может быть связана умственная отсталос ть, имеющая место у 1/3 больных ПМД Дюшетша.
Приблизительно 55-65% всех случаев ПМД Дю- шенна/Бекера обусловлены делениями гена дистрофина различной протяженности, 5-10% случаев - дупликациями части гена, у остальных больных имеют место толковые мутации [Forrest S. et al., 1988; Hu X. et al., 1988; Koenig M. et al., 1989]. Делеции в гене дистрофина распределяются отнюдь не равномерно по его длине, а преимущественно группируются вокруг двух областей гена, образуя так называемые «горячие точки» делений - в 5’-области гена (экзоны 2-20) и в его дистальной части в области экзонов 44-53 [Koenig М. et al., 1987; Forrest S. et al., 1988]. Интересно отметить, что проксимальные делеции гена чаще выявляются при семейных формах болезни, тогда как дистальные делеции обычно ассоциированы со спорадическими случаями (т.е. возникшими как результат новой мутации); при выявлении проксимальной делеции гена повторный риск заболевания в семье почти на порядок выше, чем при дистальной (соответственно, 30% и 4%) [Passos-Bueno М. et al., 1992].
Открытие генетического дефекта при ПМД Дю I пенна и Бекера дало возможность с молекулярных позиций объяснить причину различий в клинической кар- I пне этих форм миопатий. Тяжелая форма Дюшенна раз
вивается обычно при наличии мутаций, повреждающих рамку считывания кодирующей области гена либо нарушающих структуру функционально значимых доменов дистрофина [Hoffman Е. el al., 1988; Monaco A. et al., 1988; Koenig M. et al., 1989]. Результатом таких мутаций является грубое нарушение синтеза или функции дистрофина. Напротив, у больных миопатией Бекера имеют место, как правило, внутренние делеции или дупликации гена, не приводящие к сдвигу рамки считывания кодонов; вследствие этого синтезируется измененный, «усеченный» дистрофии, сохраняющий частичную функциональную активность [Hoffman Е. et al., 1988; Monaco A. et al., 1988]. He случайно у больных НМД Дюшенна отмечается полное отсутствие дистрофина при иммуногистохимическом исследовании мышечных био- птатов с помощью антидистрофиновых антител, тогда как при миопатии Бекера в препарате выявляются небольшие дистрофин-позитивные участки (рис. 33) [Hoffman Е. et al., 1988]. Указанная теория «рамки считывания», объясняющая характер клинической картины в зависимости от типа мутации, подтверждается в абсолютном большинстве (92%) всех случаев НМД Дюшен- на и Беккера [Koenig М. et al., 1989]. В зависимости от характера молекулярного дефекта могут наблюдаться клинически и иммуногистохимически «промежуточные» варианты болезни [Tinsley J. et al., 1994]; описан также ряд необычных мышечных синдромов, обусловленных мутациями гена дистрофина - крампи с рецидивирующей миоглобинурией и сниженной толерантностью к физической нагрузке, изолированная кардиомиопатия, доброкачественная миопатия позднего возраста [Doriguzzi С. et al., 1997; Palmucci L. et al., 2000]. В связи с молекулярным единством ПМД Дюшенна, ПМД Бекера и указанных выше атипичных вариантов миопатий в
литературе в последнее время принято объединять эти формы общим термином дистрофинопатии [Griggs R., Fischbeck К., 1992].
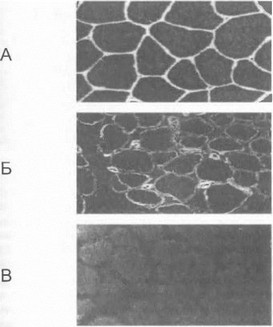
Рис. 33. Иммуногистохимнчсскип aw ал и 5 мышечных бпо- нтатов на дистрофии у бйgt;Ш*№ Г1МД Дюшспма/Бексра Л. Контроль. Б. ПМД Бекера (ха фойе нарушения нормальной окраски мышечных волокон на дистрофии выявляются отдельные днетрофпн-позиптпые участки). В. ПМД Дюшснпа (полноеотсутствие дистрофин-мо.'лтшных мышечных волокон).
В редких случаях заболевание в своем разверну- I ом виде может манифестировать у женщин, являющихся I етерозиготными носительницами мутантного гена; это наблюдается при хромосомных транслокациях и перестройках, затрагивающих критический хромосомный сегмент Хр21, при синдроме Тернера (генотип ХО), а также при несбалансированной инактивации Х-хромо- сом в раннем эмбриогенезе [Griggs R., Fischbeck К., 1992; Specht L., Kunkel L., 1993]. Во всех указанных случаях наличие мутантного гена на одной из Х-хромосом не компенсируется нормальным геном гомологичной хромосомы, что и приводит к появлению типичных симптомов болезни у женщин.
«Золотым стандартом» при постановке диагноза дистрофинопатий является исследование мышечной ткани на дистрофии (иммуногистохимический анализ, им- муноблоттинг) (рис. 33). Этот метод, однако, весьма дорог, технически сложен и требует проведения биопсии мышцы Поэтому в настоящее время широко применяется неинвазивная диагностика ПМД Дюшенна/Бекера с использованием различных молекулярно-генетических подходов на клетках крови. Наиболее простым методом ДНК-диагностики дистрофинопатий является так называемая мультиплексная (мультипраймерная) ПЦР [Chamberlain J. et al., 1988]. Она заключается в одновременной амплификации набора наиболее часто мутирующих экзонов гена дистрофина; при электрофорезе продуктов реакции отсутствие одного или нескольких экзонов будет свидетельствовать о делении, т.е. служить молекулярным подтверждением диагноза [Chamberlain J., et al., 1988; Multicenter Study Group, 1992]. Комбинированное использование нескольких мультиплексных реакций позволяет диагностировать до 98% всех делений, имеющих место у больных ПМД Дюшенна/Бекера [Beggs A. et al., 1990]. На рис. 34 показан пример прямой ДНК-диагностики мышечной дистрофии Дюшенна при мультиплексной ПЦР (выявление различных делений в гене дистрофина).
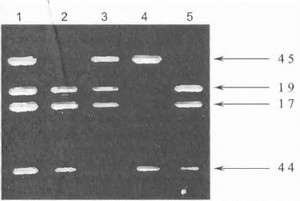
Рис. 34. Прямая ДНК-диагностика мышечной дистрофии Дюшенна с помощью мультиплексной полимеразной цепной реакции
У каждого из обследуемых лиц одноврсмеш ю амплпфицированы 4 экзона гена дистрофппа (экзоны 17,19,44 и 45; стрелки ука бывают на соответствующие продукты амплификации). Дорожка 1 - кон троль, дорожки 2-5 - больные мышечной дистрофией Дюшенна с различными делениями гена дне грофнна (дорожки 2 и 5 -деления лкзона 45, дорожка 3 - деления зкзона 44, дорожка 4 - деле! а ш эк'зо- нов 17 и 19).
Для диагностики носительства мелких или редких делеций в гене дистрофина, а также при наличии других типов мутаций (вставки и дупликации, точковые мутации) могут применяться более сложные методы ДНК-анализа, такие как Саузерн-блоттинг с использованием кДНК-зондов (см. рис. 27 на стр. 92), SSCP- и гетеродуплексный анализ отдельных экзонов гена, различные методы исследования кДНК, полученной из лимфоцитов или мышечных биоптатов с помощью обратнотранскриптазной ПЦР. а также ряд других подходов [Darras В. et al., 1988; Forrest S. et al., 1988; Roberts R. et al., 1992; Prior T. et al, 1994; Tubiello G. et al., 1995; van Essen A., 1997]. Важно отметить, что указанные ДНК- методы не только значительно повышают процент выявляемое™ мутаций в гене дистрофина у обследуемых больных, но и позволяют осуществлять диагностику мутаций у женщин-носительниц (последнее невозможно при проведении мультиплексной ПЦР, поскольку у женщин-носительниц делеция на мутантной хромосоме «маскируется» наличием нормального экзона, ампдифи- цируемого с другой Х-хромосомы).
В 25-30% всех случаев ПМД Дюшенна/Бекера у больного не удается выявить доступными методами повреждений в гене дистрофина, но при этом имеется необходимость установления генетического статуса как лиц из группы риска в обследуемой семье (сибсы мужского пола, родственники больного по женской линии), так и плода у женщин-носительниц (для решения вопроса о сохранении или прерывании беременности). В таких ситуациях может использоваться косвенная ДНК- диагностика, позволяющая проследить наследование мутантной хромосомы в изучаемой семье. Для этой процедуры необходимо наличие образцов ДНК либо заведомо больного ребенка, либо заведомо здоровых братьев пробанда. Поскольку для гена дистрофина характерна высокая частота внутригенных рекомбинаций, при проведении косвенной ДНК-диагностики ПМД Дюшенна/Бекера используются обычно несколько маркеров, расположенных в проксимальной, дистальной и центральной частях гена либо тесно сцепленных с геном и фланкирующих его с теломерного и центромерного конца [Clemens Р. et al., 1991; Feener С. et al., 1991]. На рис. 35 показан результат косвенной ДНК-диагностики ПМД Дюшенна у сибсов больного ребенка в большой семье.
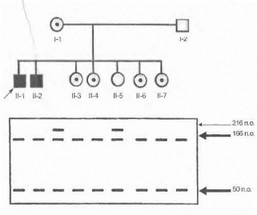
Рис. 35. Косвенная ДНК-диагностика мышечной дистрофии Дюшенна
Исследован локус pERT 87-15 (рестрикция ферментом Ват Ш). Мутантный материнский аляель визуализируется после рестрикции в виде двух фрагментов ДНК длиной 166 и 50 п.о. (жирные стрелки), нормальный материнский аллель - в виде фрагмента длиной 216 п.о. (тонкая стрелка). Дочери И 3, П-4, II-6 и 11-7 унаследовали от матери мутантный аллель маркера и являются носителями мутантной хромосомы, дочь 11-5 унаследовала от матери нормальный аллель маркера (носительство мутантной хромосомы исключено). Вероятность ошибки, связанной с возможной рекомбинацией между маркерным локусом и геном дистрофина- около 2%.
Серьезной проблемой при проведении ДНК-ди- агностики в семьях с ПМД Дюшенна/Бекера является чрезвычайно высокая частота спонтанных мутаций (10'4 на поколение), что частично может объясняться его
гигантским размером [Tinsley J. et al., 1994]. Предполагается, что около трети всех случаев ПМД Дюшенна/ Бекера обусловлены мутациями de novo [Darras В. et al., 1988]. Возникновение новых мутаций в гене дистрофи- на может происходить на любых этапах гамето- и онтогенеза, в результате чего единая клетка-предшественник дает начало смешанной популяции нормальных и мутантных клеток (феномен соматического и гонадного мо- заицизма). Возможность соматического и гонадного мозаицизма существенно видоизменяет расчеты генетического риска при проведении медико-генетическом консультирования у членов семьи, относящихся к группе риска (братья-сестры пробанда, родственники по женской линии). Более подробно этот вопрос будет рассмотрен в главе 5.