
Миотоиическая дистрофия представляет собой наиболее частую форму мышечной дистрофии взрослых (распространенность - 1 на 7500 человек) [Harper Р., 1989]. Заболевание наследуется по аутосомно-доминан- тному типу и характеризуется мультисистемностью поражения с широкой вариабельностью клинических проявлений, основными из которых являются миотония, мышечные атрофии и парезы скелетной мускулатуры (преимущесгвенно дистальных отделов конечностей и лица), катаракта, кардиомиопатия, эндокринные нарушения. В семьях с миотонической дистрофией весьма характерным является феномен антиципации, проявляющийся манифестацией более тяжелых и все более ранних форм болезни в каждом последующем поколении [Harper R, 1989].
Ген, ответственный за развитие миотонической дистрофии (DMPK), локализуется на хромосоме 19ql3.2-13.3 и кодирует синтез белка миотонинпроте- инкиназы [Brook J. et al., 1992]. Миотоническая дистрофия - яркий представитель заболевания с «динамическим» типом мутации: у больных имеет место экспансия тандемных тринуклеотидных повторов СТО (цитозин- тимин-гуанин). расположенных в З’-нетранслируемой области гена [Buxton J. et al., 1992; Fu Y. et al., 1992; Mahadevan M. et al., 1992]. Нормальные аллели гена DMPK содержат до 30 копий CTG-триплетов, тогда как на мутантных хромосомах число этих тринуклеотидных повторов увеличено в десятки и сотни раз (до 4000 CTG- повторов). Показана тесная корреляция между числом CTG-повторов и тяжестью клинического синдрома: больные с небольшой степенью экспансии триплетов (40- 160 копий) имеют минимальные клинические проявления, например - только катаракту в качестве единственного симптома [Brook J. etal., 1992; Harley Н. etal., 1993]. Напротив, больные с развернутой клинической картиной имеют большее число копий триплетов, а наибольшая степень экспансии С J G-повторов отмечена при тяжелой врожденной форме миотонической дистрофии [Lavedan С. et al., 1993]. Причиной антиципации в семьях с миотонической дистрофией является генетическая нестабильность мутантного тринуклеотидного сегмента [Harper Р. et al., 1992]. Интересно, что нестабильность CTG-повтора заметно выше при передаче гена по материнской линии [Lavedan С. et al., 1993; Mulley J. et al., 1993], поэтому врожденная форма миотонической дистрофии наблюдается исключительно при рождении детей от больных матерей. В редких случаях наблюдается уменьшение (вплоть до нормы) длины мутантного повтора у потомков, что сопровождается развитием более легкой клинической картины или асимптомным течением [Abeliovich D. et al., 1993]. Такие случаи обычно наблюдаются при отцовской передаче гена; в качестве причины обсуждается возможность селекции против больших аллелей в мужском гаметогецсзе либо неблагоприятного влияния больших аллелей на мужскую фертильность [Harley Н. et al., 1993; Lavedan С. et al., 1993].
Предполагается, что экспансия тринуклеотидных I ювторов при миотонической дистрофии может обусловливать нарушение транскрипции/трансляции, либо влиять на стабильность соответствующей мРНК, резудьта- I ом чего является угнетение синтеза миотонинпротеин- киназы [Harley Н. et al., 1993.; Willems Р., 1994]. Другим гипотетическим механизмом реализации данной мутации может быть негативное влияние патологически удлиненного тринуклеотидного сегмента на экспрессию некоторых близлежащих генов в локусе 19q 13 [Willems Р, 1994; Klesert Т. et al., 1997].
Непосредственное определение мутантного аллеля гена миотонической дистрофии путем электрофоретического разделения ПЦР-продуктов может проводиться только у больных с небольшой степенью экспансии CTG-повторов (50-100 триплетов); эффективность амплификации более длинных и сверхдлинных мутантных аллелей гена DMPK недостаточна для непосредственной визуализации продуктор ПЦР при электрофорезе. Поэтому чаще всего для прямой ДНК-диагностики миотонической дистрофии используется блот-гибридизация рестрицированной геномной ДНК или продуктов амплификации с (CTG)n- зондами [Harley Н. et al., 1993; Lavedan С. et al., 1993]. При такой диагностике мутантный аллель нередко визуализируется как смазанный протяженный след на радиоавтографе (рис. 39), что является отражением особенностей амплификации сверхдлинных тринуклеотидных аллелей либо следствием соматического мозаицизма мутантного CTG-повтора в клетках крови [Lavedan С. et al., 1993]. Нами совместно с Институтом молекулярной генетики РАН проведена прямая ДНК-диагностика болезни в 30 российских семьях с миотонической дистрофией: экспансия тринуклеотидных CTG-повторов в одном из аллелей гена у обследованных больных выявлена более чем в 90% семей, при этом величина экспансии повторов составила от 130 до 1500 CTG-копий (рис. 39).
Выявление нами нормальной длины тринуклео- тидного сегмента гена у небольшой части обследованных больных миотонической дистрофией соответствует наблюдениям других авторов [Thornton С. et al., 1994] и может быть обусловлено либо казуистическими случая ми точковых мутаций в гене миотонинпротсинкиназы [Harley И. et al., 1993], либо существованием других ло кусов, ассоциированных с данным клиническим синдромом
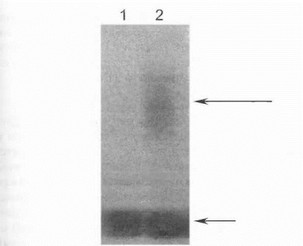
1’ис. 39. Прямая ДНК-диагностика миотонической дистро- (|ши с помощью блог-гибридизации поСаузерну Дорожка 1 - отсутствие мутации (нормальная длина обоих аллелей I сна миотонинпротеинкиназы), дорожка 2- больной миотонической дистрофией (экспансия трннуклсотндных CTG-повторов в одном из аллелей гена). Мутантный аллель обозначен длинной стрелкой, нормальный аллель - короткой стрелкой.
Г енетическая гетерогенность миотонической дистрофии была подтверждена в 1998 году: второй локус «классического» дистального фенотипа миотонической дистрофии был картирован на хромосоме 3q [Ranum L. et ale, 1998]. Более того, в том же участке хромосомы 3q I неположен ген особого и чрезвычайно редкого заболела пия - так называемой проксимальной миотонической миопатии(РКОММ) [Ricker К. etal., 1999]. Предполагайся, что указанные клинические синдромы (второй ва- I н 1ант дистальной миотонической дистрофии и прокси
мальная миотоническая миопатия) могут являться аллельными заболеваниями, обусловленными мутациями одного гена [Thornton С., AshizawaT., 1999], однако подтверждение этой гипотезы станет возможным лишь после идентификации данного гена на хромосоме 3q.
Ген, ответственный за развитие миотонической дистрофии (DMPK), локализуется на хромосоме 19ql3.2-13.3 и кодирует синтез белка миотонинпроте- инкиназы [Brook J. et al., 1992]. Миотоническая дистрофия - яркий представитель заболевания с «динамическим» типом мутации: у больных имеет место экспансия тандемных тринуклеотидных повторов СТО (цитозин- тимин-гуанин). расположенных в З’-нетранслируемой области гена [Buxton J. et al., 1992; Fu Y. et al., 1992; Mahadevan M. et al., 1992]. Нормальные аллели гена DMPK содержат до 30 копий CTG-триплетов, тогда как на мутантных хромосомах число этих тринуклеотидных повторов увеличено в десятки и сотни раз (до 4000 CTG- повторов). Показана тесная корреляция между числом CTG-повторов и тяжестью клинического синдрома: больные с небольшой степенью экспансии триплетов (40- 160 копий) имеют минимальные клинические проявления, например - только катаракту в качестве единственного симптома [Brook J. etal., 1992; Harley Н. etal., 1993]. Напротив, больные с развернутой клинической картиной имеют большее число копий триплетов, а наибольшая степень экспансии С J G-повторов отмечена при тяжелой врожденной форме миотонической дистрофии [Lavedan С. et al., 1993]. Причиной антиципации в семьях с миотонической дистрофией является генетическая нестабильность мутантного тринуклеотидного сегмента [Harper Р. et al., 1992]. Интересно, что нестабильность CTG-повтора заметно выше при передаче гена по материнской линии [Lavedan С. et al., 1993; Mulley J. et al., 1993], поэтому врожденная форма миотонической дистрофии наблюдается исключительно при рождении детей от больных матерей. В редких случаях наблюдается уменьшение (вплоть до нормы) длины мутантного повтора у потомков, что сопровождается развитием более легкой клинической картины или асимптомным течением [Abeliovich D. et al., 1993]. Такие случаи обычно наблюдаются при отцовской передаче гена; в качестве причины обсуждается возможность селекции против больших аллелей в мужском гаметогецсзе либо неблагоприятного влияния больших аллелей на мужскую фертильность [Harley Н. et al., 1993; Lavedan С. et al., 1993].
Предполагается, что экспансия тринуклеотидных I ювторов при миотонической дистрофии может обусловливать нарушение транскрипции/трансляции, либо влиять на стабильность соответствующей мРНК, резудьта- I ом чего является угнетение синтеза миотонинпротеин- киназы [Harley Н. et al., 1993.; Willems Р., 1994]. Другим гипотетическим механизмом реализации данной мутации может быть негативное влияние патологически удлиненного тринуклеотидного сегмента на экспрессию некоторых близлежащих генов в локусе 19q 13 [Willems Р, 1994; Klesert Т. et al., 1997].
Непосредственное определение мутантного аллеля гена миотонической дистрофии путем электрофоретического разделения ПЦР-продуктов может проводиться только у больных с небольшой степенью экспансии CTG-повторов (50-100 триплетов); эффективность амплификации более длинных и сверхдлинных мутантных аллелей гена DMPK недостаточна для непосредственной визуализации продуктор ПЦР при электрофорезе. Поэтому чаще всего для прямой ДНК-диагностики миотонической дистрофии используется блот-гибридизация рестрицированной геномной ДНК или продуктов амплификации с (CTG)n- зондами [Harley Н. et al., 1993; Lavedan С. et al., 1993]. При такой диагностике мутантный аллель нередко визуализируется как смазанный протяженный след на радиоавтографе (рис. 39), что является отражением особенностей амплификации сверхдлинных тринуклеотидных аллелей либо следствием соматического мозаицизма мутантного CTG-повтора в клетках крови [Lavedan С. et al., 1993]. Нами совместно с Институтом молекулярной генетики РАН проведена прямая ДНК-диагностика болезни в 30 российских семьях с миотонической дистрофией: экспансия тринуклеотидных CTG-повторов в одном из аллелей гена у обследованных больных выявлена более чем в 90% семей, при этом величина экспансии повторов составила от 130 до 1500 CTG-копий (рис. 39).
Выявление нами нормальной длины тринуклео- тидного сегмента гена у небольшой части обследованных больных миотонической дистрофией соответствует наблюдениям других авторов [Thornton С. et al., 1994] и может быть обусловлено либо казуистическими случая ми точковых мутаций в гене миотонинпротсинкиназы [Harley И. et al., 1993], либо существованием других ло кусов, ассоциированных с данным клиническим синдромом
1’ис. 39. Прямая ДНК-диагностика миотонической дистро- (|ши с помощью блог-гибридизации поСаузерну Дорожка 1 - отсутствие мутации (нормальная длина обоих аллелей I сна миотонинпротеинкиназы), дорожка 2- больной миотонической дистрофией (экспансия трннуклсотндных CTG-повторов в одном из аллелей гена). Мутантный аллель обозначен длинной стрелкой, нормальный аллель - короткой стрелкой.
Г енетическая гетерогенность миотонической дистрофии была подтверждена в 1998 году: второй локус «классического» дистального фенотипа миотонической дистрофии был картирован на хромосоме 3q [Ranum L. et ale, 1998]. Более того, в том же участке хромосомы 3q I неположен ген особого и чрезвычайно редкого заболела пия - так называемой проксимальной миотонической миопатии(РКОММ) [Ricker К. etal., 1999]. Предполагайся, что указанные клинические синдромы (второй ва- I н 1ант дистальной миотонической дистрофии и прокси
мальная миотоническая миопатия) могут являться аллельными заболеваниями, обусловленными мутациями одного гена [Thornton С., AshizawaT., 1999], однако подтверждение этой гипотезы станет возможным лишь после идентификации данного гена на хромосоме 3q.