
Аутосомные формы конечностно-поясных ПМД (в старой номенклатуре - ПМД Эрба-Рота) являются относительно редкими, их суммарная распространенность составляет приблизительно 50 случаев на миллион населения [Emery А., 1991].
Клиническая картина конечностно-поясной ПМД (КПМД) характеризуется началом болезни с мышц тазового пояса и бедер, с последующим вовлечением мускулатуры плечевого пояса и проксимальных отделов рук и постепенной генерализацией процесса [Shields R., 1994; van der Kooi A. et al., 1994]. Диагноз КПМД может быть поставлен только после исключения многочисленных гено- и фенокопий, таких как митохондриальные, вос- папительные, эндокринные, метаболические миопатии дистрофинопатии, спинальные амиотрофии и др. [Bushby К., 1995]. КПМД может передаваться как до аутосомно-доминантному, так и по аутосомно-рецессив- ному типу (соответственно, КПМД] и КПМД2). Значительно более частым является аутосомно-рецессивное наследование, при котором заболевание нередко проявляется в семье как единичный (спорадический) случай.
Аутосомно-рецессивные КПМД. Конечностно- поясной вариант мышечной дистрофии с аутосомно-ре- цессивным типом наследования характеризуется выраженной генетической гетерогенностью. К настоящему времени известно сущестовапие, как минимум, 9 локусов данных заболеваний, причем для 7 форм идентифицированы гены и их белковые продукты [Иллариошкин
С.Н., Иванова-Смоленская ИА., 1998; Weiler Т. et al., 1998; Bushby К., 1999; Driss A. et al., 2000]. В соответствии с современной геномной классификацией, аутосомно-рецессивные мышечные дистрофии (КПМД2) и соответствующие хромосомные локусы обозначаются буквенными символами в порядке их открытия - К11МД2И, 25, 2С, 2D и т.д. (см приложение 1). Знание первичных молекулярных дефектов при большинстве форм аутосомно-рецессивных КПМД позволяет подразделить их на 3 большие группы: саркогдиканопатии, кальпаинопатии и дисферлинонатии.
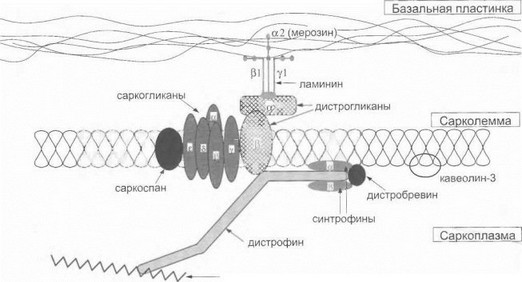
К саркогликстопатиям относятся мышечные дистрофии, обусловленные мутациями в генах особых с сруктурных белков мышечной мембраны - саркоглика- нов [Tinsley .1. etal., 1994; Ozawa Е. etal., 1995; Worton R., 1495]. Саркоглитсаны являются элементами сложного ылкового комплекса мышечного волокна, состоящего из шетрофина и дистрофин-ассоциированных протеинов саркогликанов, дистрогликанов, синтрофинов, дисг- цобревина, мерозина и др. (рис. 36).
Различные компоненты данного белкового комп- чскса (обозначаемого как дистрофии-гликопротеиповый
F-актин
Рис. 36. Струкаура дистрофин-гликопротеинового комплекса
146 Глава 3
комплекс) образуют единую «ось», обеспечивающую эластичную связь между филаментами актина, сарколеммой и внеклеточным матриксом. Считается, что такая структурно-функциональная организация дистрофин- гликопротеинового комплекса способствует фиксации миоцита к элементам внеклеточного матрикса и стабилизирует мышечную мембрану, предохраняя ее от повреждения в процессе мышечных сокращений [Campbell К., 1995; Ozawa Е. etal., 1995]. Ключевое значение в стабилизации всего дистрофин-гликопротеино- вого комплекса придается дистрофину, поэтому у больных ПМД Дюшенна/Бекера имеют место вторичные изменения дистро- и саркогликанов мышечной мембраны [Tinsley J. et al., 1994]. Первичные саркогликанопатии связаны непосредственно с патологией этих белков, тогда как уровень и локализация дистрофина в мышечном волокне могут оставаться нормальными. Известны 4 формы аутосомно-рецессивных КПМД, относящихся к первичным саркогликанопатиям: 1) КПМД2С, обусловленная мутациями гена у-саркогликана на хромосоме 13q 12 [Noguchi S. et al., 1995]; 2) КПМД2Д обусловленная мутациями гена а-саркогликана (адгалина) на хромосоме 17q12-21.33 [Roberds S. etal., 1994]; 3) Ю1МД22ц вызываемая мутациями гена Р-саркогликана на хромосоме 4ql2 [Lim L. et al., 1995]; 4) КИМД2Т', связанная с мутациями гена 5-саркогликана на хромосоме 5с]33—34 | Nigro V. et al., 1996]. При всех формах саркогликанопа- тий отмечается копечпостно-пояспой тип распределения парезов скелетных мышц, тогда как тяжесть клинической картины определяется характером молекулярного дефекта. При миссенс-мутацчях с заменой аминокислоты заболеванию обычно свойственно позднее начало (на 2-3-м десятилетии жизни) и сравнительно благоприятное течение [Lim L. et al., 1995; Piccolo ff. et al., 1995]; напротив, при дефектах, грубо нарушающих синтез белка (деледии со сдвигом рамки, нонсенс-мутации), имеет место ранний дебют заболевания и быстрое прогрессирование - так называемая «тяжелая детская аутосомно- рецессивная мышечная дистрофия», иди «дюшенно-по- добная мышечная дистрофия» [Campbell К., 1995; Lim L. et al., 1995; Piccolo F. et al., 1995]. Клинически дифференциальная диагностика саркогликанопатий с ПМД Дюшенна/Бекера как правило представляет собой весьма непростую задачу, особенно учитывая тот факт, что для саркогликанопатий достаточно характерны задержка раннего двигательного развития, псевдогипертрофии икроножных мышц, в некоторых случаях - кардиомиопатия [Bushby К., 1999].
К калъпаинолатии относится форма КП МД 2/1, обусловленная мутациями в гене на хромосоме 15ql5, кодирующем синтез мышечно-специфичного фермента кальлаина-З [Richard I. et al., 1995]. Данный фермент принадлежит к семейству внелизосомных внутриклеточных цистеиновых протеаз; предположительно, его функции могут быть связаны с регуляцией процессов диф- ференцировки мышечной ткани и влиянием на экспрессию факторов транскрипции [Wang К. et al., 1989]. В отличие от саркогликанопатий, для КПМД2д характерна тенденция к более позднему началу болезни и более медленному прогрессированию, нормальное физическое развитие в раннем детстве, длительно наблюдающееся преобладание слабости в мышцах-аддукторах бедра по сравнению с абдукторами, отсутствие мышечных псевдогипертрофий и кардиомиопагии [Bushby К., 1999]. Все эти признаки не могут, однако, служить абсолютно надежной основой для клинической диагностики данной формы К11МД.
К дисферлинопатиям относится большое число аллельных вариантов аутосомно-рецессивной ПМД, обусловленных мутациями в гене дисферлина на хромосоме 2р 13 [Bashir R. et al., 1994; 1998; Bejaoui К. et al., 1995; Passos-Bueno M. et al., 1995; Liu J. et al., 1998; Weiler T. et a]., 1999]. Дисферлин представляет собой новый белок, ассоциированный с мышечной мембраной, функции которого окончательно не установлены; высказывается предположение, что дисферлин может участвовать в процессах регуляции слияния миобластов [Bashir R. et al., 1998; Liu J. et al., 1998]. Для дисферли- нопатий характерен чрезвычайно широкий фототипический полиморфизм. Чаще всего они манифестируют в виде конечностно-ноясного варианта мышечной дистрофии (К1гМД25), особенностями которого являются нормальное раннее развитие, начало болезни в конце 2-го десятилетия жизни, относительно «мягкое» течение, преобладание слабости в задних группах мышц ног (особенно при распространении процесса дистально), чрезвычайно высокий уровень сывороточной креатинфосфо- киназы (gt;50-100 раз выше нормы) [Passos-Bueno М. et al., 1995; Bushby К., 1999]. Аллельными вариантами дис- ферлинопатий являются также некоторые редкие формы дистальных ПМД - миопатия Миоши (характеризующаяся слабостью и атрофией дистальных отделов ног, преимущественно - икроножной мышцы) и тибиальная дистальная миопатия [Miyosh; К etal., 1986; Bashir R et ah, 1998; Liu.I. etal., 1998]. Более того, в литературе представлены наблюдения двух уникальных инбредных родословных (одна из них описана нами в дагестанском горном изоляте, вторая - канадскими исследователями в изоляге индейцев Манитобы), в которых у родственников с идентичными мутациями в гене дисферлина имела место либо КПМД25, либо миопатия Миоши |Illarioshkm S. et al., 1996; Weiler T. et al., 1996; 1999]. Данный пример показывает, что отнесение различных фенотипических вариантов ПМД к группе дисферлино- патий только по клиническим данным невозможно и должно основываться на молекулярных методах исследования.
Совсем недавно, в 2000 году, был идентифицирован еще один генетический дефект в группе аутосомно- рецессивных конечностно-поясных мышечных дистрофий, ответственный за развитие КПМД2С: данный молекулярный вариант болезни обусловлен мутациями в гене саркомерного белка телетонина (хромосома 17ql 1—12) [Moreira Е. et al., 2000].
Ведущее значение в дифференциальной диагностике аутосомно-рецессивных КПМД имеет определение типа наследования болезни, а также иммуногистохими- ческий анализ биоптатов мышц с использованием антител к соответствующим белкам. У больных с сарган ли- канопатиями окраска на дистрофии остается нормальной; в то же время дефект любого из белков-саркоглика- нов обычно приводит к дезинтеграции всего саркогли- канового комплекса и нарушению нормального окрашивания препарата при использовании антисаркогликано- вых антител (рис. 37) [Ozawa Е. et al., 1995]. Аналогичным образом другие молекулярные формы аутосомно- рецессивных мышечных дистрофий диагностируются с помощью выявления специфического белкового дефекта кальпаина-3 или дисферлина в мышечном препарате [Anderson L. et al., 1998; Matsuda C. et al., 1999]. После того как иммуногистохимический анализ позволил определить потенциальный ген-кандидат, может ставиться вопрос о проведении мутационного скрининга кодирующей области данного гена, обнаружении конкретных мутаций и проведении прямой ДНК-диагностики у лиц из группы риска [Beckmann J., Bushby К., 1996; Duggan
- et al., 1997; Bushby K., 1999]. Принимая во внимание большие размеры гепов аутосомно-рецессивных КПМД и разнообразие описанных му гаций, прямая ДНК-диаг- ностика этих заболеваний до сегодняшнего времени представляет серьезные трудности и может проводиться лишь в небольшом числе высокоспециализированных лабораторий.
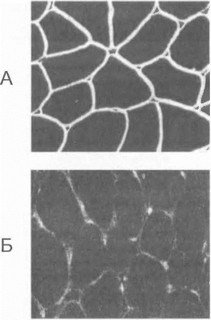
Рис. 37. Иммуногистохнмичсекпй анализ мышечных био- I патов на сх-саркогликап у больного KI1МД2/)
Л Контроль. Б. Конечиостно-поясная мышечная дистрофия типа W (нарушение нормальной окраски мышечных волокон на а -сар- когликан).
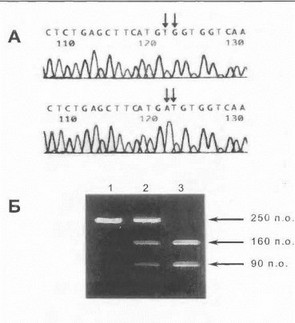
Рис 38. Прямая ДНК-диагностика дисферлинопагии в семье с KI1МД2Д и миопатией Миоши
А. Точковая мутация TG573/574AT в гомозиготном состоянии (фра! мент нуклеотидной последовательности экзона ЗтенЛ дисфсрлина). Мутантные нуклеотиды указаны стрелками. Б. Рестрикционный анализ продуктов амплификации экзона 3 с использованием фермента Rea 1. Мутация TG573/574AT создает новый сайт рестрикции для Rea I. Нормальный аллель имеет размер 250 и.о., мутантный аллель разрезается на фрагменты 160 и 90 п.о. Дорожка 1 - контроль, дорожка 2 - гетерозиготный носитель мутации 573-574TG—эАТ, дорожка 3 - гомозиготный носитель мутации 573-574TG—эАТ (больной).
В нашей стране впервые прямая ДНК-диагностика отдельных случаев аутосомно-рецессивных КПМД была проведена двумя группами исследователей - нейрогене- тическим отделением НИИ неврологии РАМН в семье с КПМД2В и миопатией Миоши (рис. 38) и сотрудниками Медико-генетического научного центра РАМН и Института молекулярной генетики РАН в семьях с КПМД2П [Липатова Н.Л. и др., 2000]. В достаточно больших информативных семьях предпочтительным подходом является косвенная ДНК-диагностика болезни (анализ генетического сцепления с одним из локусов на хромосомах 2, 4, 5, 9, 13, 15, 17 и 19) [Beckmann J., Bushby К., 1996; Weiler Т. et al., 1998; Bushby К., 1999].
Комбинированное использование методов белковой и генной диагностики позволило оценить сравнительную частоту конкретных форм аутосомно-рецессив- ных мышечных дистрофий: в большинстве изученных популяций мира не менее 40% больных с КПМД имеют первичную патологию кальпаина-3, около 10-25% больных страдают дисферлинопатиями и различными вариантами саркогликанопатий (из них чаще всего встречается К11МД2Д связанная с патологией а-саркогликана), тогда как все остальные формы аутосомно-рецессивных КПМД являются более редкими [Bushby К., 1999]. В отдельных популяциях эти цифры могут существенно различаться, причем знание о преобладании в той или иной популяции определенной молекулярной формы мышечной дистрофии (и даже определенной мутации) позволяет значительно упростить идентификацию повреждений в гене и проведение ДНК-диагностики в семьях соответствующего этнического происхождения.
Аутосомно-доминантные КПМД. Семьи с ауто- сомно-доминантным наследованием являются весьма редкими среди всех случаев КПМД (не более 10%) [Bushby К., 1999]. Общими клиническими особенностями данных заболеваний являют ся достаточно выраженная вариабельность возраста начала болезни, незначительное повышение уровня сывороточной креатинфос- фокиназы (обычно в 4-6 раз, нередко может быть в норме), а также сравнительно медленное прогрессирование. Важно отметить, что примерно у 8% пациентов с клиническим диагнозом аутосомно-доминантной КПМД может выявляться генетический дефект, характерный для лице-лопаточно-плечевой мышечной дистрофии (см. раздел 3.1.1.3), поэтому исключение последней формы ПМД с помощью ДНК-диагностики должно быть первым шагом при обследовании таких больных [van der Kooi A. et al., 1994].
На сегодня известны 5 различных генетических локусов аутосомно-доминантных КПМД - на хромосомах 5q (форма КПМД7Н), lqll-21 (КПМД7Д), Зр25 (КПМД 1C), 6q22 (КПМД 1D) и 7q (КТТМД7А) [Speer М. et al., 1999; Messina D. et al., 1997; Minetti C. et al., 1998; Hauser M. et al., 2000; Muchir A. et al., 2000]. Первичные молекулярные дефекты при 3 формах идентифицированы. Форма КПМД 1А обусловлена мутациями структурного саркомерного белка мышечного волокна миотили- на [Hauser М. et al., 2000]. Форма К11МД7С вызывается мутациями в гене мышечного белка кавеолина-3, выполняющего сигнальные регуляторные функции [McNally Е. et al., 1998; Minetti С. et al., 1998]; предполагается, что кавеолин-3 может иметь отношение к регуляции процессов гликолиза в скелетной мышце [McNally Е. et al., 1998]. Форма КПМД77? является аллельной с аутосомно-доминантной мышечной дистрофией Эмсри-Дрейфуса (см. раздел 3.1.1.6) и обусловлена мутациями гена ламина АУС, кодирующего белки ламины-компоненты ядерной мембраны [Muchir A. et al., 2000].
До настоящего времени в мире опыт ДНК-диагностики аутосомно-доминантных КПМД весьма невелик и ограничивается, главным образом, возможностью кос-
ионного ДНК-тестирования в информативных семьях, в которых подтверждено генетическое сцепление с одним пз указанных локусов. В семьях с К ПМД УД, Ю 1МДУ/У и К11МД7С в случае обнаружения мутаций в генах миоти- лина, ламина А/С и кавеолина-3 возможно проведение прямой ДНК-диагностики у лиц из группы риска.