
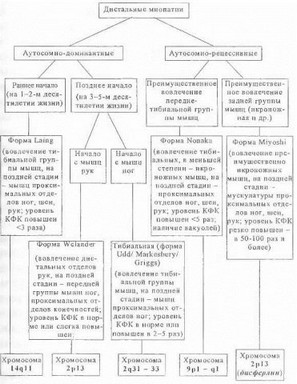
Дистальные ПМД (дистальные миопатии). Дистальные миопатии представляют собой клинически и генетически гетерогенную группу заболеваний, характеризующихся избирательным или преимущественным поражением дистальной мускулатуры нижних и/или верхних конечностей. В соответствии с типом наследования, возрастом начала болезни и локализацией мышечных атрофий и парезов дистальные миопатии подразделяются на несколько самостоятельных клинических форм [Griggs R., Markesbery W., 1994; Mastaglia F., LaingN., 1999]. Современная классификация основных форм дистальных миопатий представлена па рис. 40.
Как видно на этой схеме, наиболее четко тип дистальной миопатии дифференцируется в начальной стадии болезни, когда возможно определение паттерна вовлечения скелетной мускулатуры «в чистом виде». С годами, по мере прогрессирования заболевания при большинстве форм в основном сохраняется относительная селективность поражения соответствующих групп мышц, однако наблюдается постепенное распространение процесса на более проксимальные отделы и ишэгда - противоположные конечности и мускулатуру шеи. Например, на поздней стадии Чкистевой миопатии» Веландер может наблюдаться вовлечение мышц ног, а при остальных формах дистальных миопатий (за исключением ти- биальной миопатии Удда) заболевание, начавшись с дистальной мускулатуры ног, может распространяться на
Гис. 40. Современная клинико-генетическая классификация дистальных мйопатий
верхние конечности [Mastaglia R, Laing N., 1999]. Важным дифференциально-диагностическим признаком является преимущественное вовлечение передней или задней группы мышц ног: так, для миопатии Миоши наи- f юлее характерным признаком (наряду со значительно
повышенным уровнем сывороточной креатинфосфоки- назы) является более грубое поражение икроножной мышцы и связанные с этим затруднения при ходьбе на носках, тогда как при большинстве других форм дистальных миопатий наблюдается преимущественное поражение передней (тибиальной) группы мышц с невозможностью ходьбы на пятках. У больных дистальными мио- патиями Нонака и Веландер при биопсии пораженных мышц, помимо типичных дистрофических изменений, выявляются кольцевые вакуоли и тубулофиламентозные включения, что сближает данные формы дистальных миопатий с редким аутосомно-рецессивным заболеванием скелетных мышц - наследственным миозитом с включениями [Mitrani-Rosenbaum S. etal., 1996].
На сегодняшний день известна хромосомная локализация 6 генов дистальных миопатий (см. рис. 40), однако лишь для миопатии Миоши идентифицировап сам ген и его молекулярный продукт - белок дисферлин [Liu J. et al., 1998]. Как указывалось в разделе 3.1.1.2, мутации в гене дисферлина могут приводить к манифестации не только миопатии Миоши, но и другой формы аутосомно-рецессивной мышечной дистрофии - КПМД2.8. Принимая во внимание тот факт, что ген дисферлина включает 55 экзонов и имеет длину кодирующей области около 6,9 кб, поиск мутаций в данном гене и прямая ДНК-диагностика миопатии Миоши весьма трудоемки и на практике редко осуществимы. Для молекулярной диагностики миопатии Миоши более удобным является стандартный для всех ПМД подход, основанный на выявлении характерного белкового дефекта при иммуногистохимическом исследовании биоптатов мышц с помощью антител к дисферлину [Matsuda С. et al., 1999]. Для всех других форм дистальных миопатий может ставиться вопрос о проведении косвенной ДНК- диагностики на основе анализа генетического сцепления с локусами 2р 13, 2q31-33, 9р 1 -q 1 и 14q 11 [Mastaglia F., LaingN., 1999]. Однако установление сцепления (т.е. получение диагностически значимого Лод- балла) осуществимо лишь в очень небсщыном числе семей с несколькими больными родственниками, а правильно выбрать необходимый для исследования хромо- ¦ сомный локус только на основании клинической картины не представляется возможным. Поэтому в целом косвенная ДНК-диагностика дистальных миопатий имеет пока весьма ограниченное значение. В будущем, после идентификации генов дистальных миопатий и их белковых продуктов будет возможной стандартная иммуноги- стохимическая диагностика болезни и прямое определение мутаций у больных лиц и их родственников из группы риска.
Окулофарингеальная ПМД. Окулофарингеальная форма ПМД представляет собой миодистрофию позднего возраста, обычно манифестирующую на 6-м десятилетии жизни. В большинстве случаев тип наследования болезни аутосомно-доминантный; описаны единичные семьи с аутосомно-рецессивной передачей окулофарин- I сальной ПМД [Tome F., Fardeau М., .1994]. Заболевание клинически характеризуется развитием прогрессирующей слабости и атрофий проксимальных отделов конечностей, расстройствами глотания и фонации, птозом, нарушением движений глазных яблок и (на поздней стадии) слабостью лицевой мускулатуры, а патоморфоло- I п чески-миодистрофическими изменениями указанных I рупп скелетных мышц и появлением патогномоничных фпламентозных внутриядерных включений в мышечных волокнах [Tome F., Fardeau М., 1994; Emery А., 1998].
Ген окулофарингеальной ПМД на хромосоме I q 11.2—13 ответственен за синтез ядерного белка I V15Р2, служащего фактором полиаденилирования мРНК | Krais В. et al., 1998]. У всех больных окулофарингеаль- ной ПМД в мутантном гене обнаруживается увеличение числа копий тринуклеотидных повторов GCG в 1-м эк- зоне гена: в норме ген содержит 6 тандемных копий повторов GCG, кодирующих полиаланиновый участок в N- терминальной области белка, тогда как у больных число повторов в мутантном гене увеличено до 8-13 копий [BraisB.etal., 1998;Grewal R. etal., 1999; MirabellaM. et al., 2000]. Предполагается, что удлинение полиаланино- вого участка белка РАВР2 приводит к олигомеризации мутантных белковых молекул, что и лежит в основе образования внутриядерных филаментозных включений. В нормальной популяции у 2% лиц на одной из хромосом обнаруживается «промежуточный» аллель гена, имеющий 7 повторов GCG, наличие которого в гетерозиготном состоянии не сопровождается каким-либо симптомами. Однако у гомозиготных носителей аллеля (GCG) развивается клиника окулофарингеальной ПМД, причем в таких семьях имеет место аутосомно-рецессивный тип наследования болезни [Brais et al., 1998]. Гомозиготность по более длинным GCG-повторам (т.е. двойная доза заведомо мутантного аллеля) характеризуется развитием более тяжелой клинической картины и более ранним дебютом симптомов (в среднем на 18 лет раньше чем у гетерозиготных носителей мутации) [Blumen S. et al., 1999]. Таким образом, с точки зрения мутационного механизма окулофарингеальная ПМД является уникальным заболеванием с двух точек зрения: во-первых, при этой форме миодистрофии имеет место наиболее короткая экспансия повторов по сравнению со всеми другими «тринуклеотидными» заболеваниями; во вторых, различная по тяжести, но одинаковая по характеру мутация в одном и том же гене может служить причиной развития как аутосомно-доминантной, так и аутосомно-рецессив- ной формы болезни.
Открытие гена РАВР2 и универсального типа мутации в нем позволяет проводить сравнительно простую и достоверную прямую ДНК-диагностику окуло- фарингеальной мышечной дистрофии, основанную на амплификации тринуклеотидного участка гена с помощью ПЦР. Подтверждением диагноза служит обнаружение у больного на электрофореграмме двух различных фрагментов ДНК, один из которых имеет нормальный размер (6 повторов GCG), а другой - патологически удлинен и соответствует экспансии GCG-повторов gt;8 копий. Вывление «промежуточного» ПЦР-продукта с 7 повторами GCG является диагностически значимым в том случае, если оба аллеля имеют такой размер либо второй аллель является заведомо мутантным (gt;8 повторов GCG).
ПМД Эмери-Дрейфуса. Мышечная дистрофия Эмери-Дрейфуса относится к редким формам ПМД и имеет ряд специфических особенностей клинической картины: болезнь начинается в детском или юношеском возрасте, характеризуется относительно медленным прогрессированием миодистрофии (или даже стационарным течением), ранним развитием типичных контрактур в Локтевых, голеностопных и межпозвонковых суставах, а также поражением проводящей системы сердца [Emery А., 1998]. Тип наследования в абсолютном большинстве случаев - Х-сцепденный рецессивный. Ген заболевания локализован на хромосоме Xq28 и кодирует белок с неустановленной функцией эмерин [Bione et al 1994]. Эмерин экспрессируется в клетках мышечной и цругих тканей, локализуется наядерной мембране и, возможно, принимает участие в регуляции экспрессии генов [Manilal S. et al., 1996]. При ПМД Эмери-Дрейфуса описано более 60 различных мутаций в гене эмерина | Yates J., Wehnert М., 1999]. Идентификации мутации у конкретного больного предполагает проведение скрининга всей кодирующей области гена, поэтому прямая ДНК- диагностика болезни на сегодня не является рутинной и может проводиться лишь в высокоспециализированных лабораториях. Более доступным молекулярным диагностическим тестом является обнаружение дефекта белка эмерина в биоптатах мышц при иммуногистохимичес- ком анализе и Вестерн-блоттинге [Manilal S. et al., 1996; Nagano A. et al., 1996].
В единичных семьях описаны аутосомно-доми- натный и аутосомно-рецессивный варианть. ПМД Эмери-Дрейфуса. Оба они обусловлены различными по тяжести мутациями в гене на хромосоме 1 q21.3 (LMNA), кодирующем белки внутренней поверхности ядериой мембраны - ламины А/С, тесно связанные с эмерином | Bonne G. et al., 1999; Di BarlettaM. et al., 2000]. Очевидно, что мутации в данном гене могут иметь различную пежг.грантность и действовать как доминантные (вызывая болезнь в гетерозиготном состоянии) или как рецессивные (не проявляясь у гетерозигот и приводя к патологии мышц только при наличии у больного двух мутантных хромосом). По мнению М. Di Barletts с соавторами (2000), поиск мутаций в гене LMNA следует проводить у всех больных с ранними контрактурами плече- перонеальных мышц и/или синдромом «ригидного позвоночника» даже при отсутствии типичного развернутого фенотипа ПМД Эмери-Дрейфуса.
Как уже указывалось выше, аутосомные формы ПМД Эмери-Дрейфуса являются аллельными заболеваниями с КПМД типа 1В.
Миопатия Бетлема. Миопатия Бетлема представляет собой редкую аутосомно-доминантную форму ПМД, которая некоторыми авторами относится к группе врожденных миопатий. Заболевание характеризуется манифестацией симптомов в раннем детском возрасте, доброкачественным течением с практически нормальной продолжительностью жизни, конечностно-поясным типом распределения мышечных атрофий, сохранностью лицевых мышц. Важнейшим диагностическим признаком миопатии Бетлема является развитие ранних (нередко врожденных) контрактур в межфаланговых, локтевых и голеностопных суставах [Mohire М. et al., 1988]. Данная форма мышечной дистрофии является генетически гетерогенной. Оба известных гена миопатии Бетлема, локализованные на хромосомах 21q22.3 и 2q37, кодируют синтез различных субъединиц (ос1/ос2 и аЗ) коллагена VI типа - белка скелетных мышц, обеспечивающего связь базальной пластинки с гликопротеинами внеклеточного матрикса [Jobsis G. et al., 1996]. К настоящему времени в мире в нескольких десятках семей получен опыт прямой ДНК-диагиостики миопатии Бетлема на основе мутационного скрининга генов коллагена VI.