
Наследственные формы невропатий составляют до 60-70% всех хронических полиневропатий в различных популяциях и встречаются с частотой не менее 1 на 2500 [Emery А., 1991]. Данная группа заболеваний весьма гетерогепна. Клиническая классификация наследственных невропатий предполагает подразделение их на 3 основные группы: 1) наследственные моторно-сенсорные невропатии; 2) наел еде гвенные моторные невропатии; 3) наследственные сенсорные и сенсорно-вегетативные невропатии. Данное деление основано на характере симптомов и типе волокон периферических нервов, преимущественно вовлекаемых в дегенеративный процесс |Dyek Р. et al., 1993] Имеется также ряд других редких форм наследственных невропатий, многие из которых являются проявлением различных системных генетических нарушений обмена (амилоидные невропатии, пор- фирийные невропатии, некоторые метаболические лей- кодистрофии и др.).
Наследственные моторно-сенсорные невропатии (I iMCH) являются самыми распространенными в рассматриваемой группе заболеваний и имеют наибольшее значение в клинической практике [Dyck Р. et al., 1993; ('liance Р., Fischbeck К., 1994]. Большинство заболеваний из группы НМСН имеют весьма сходные клинические проявления и на протяжении многих десятилетий обозначались как «болезнь Шарко-Мари-Тута» (Charcot-Marie-Tooth) [Harding А., 1995J. Характерным для них являются развитие хронически прогрессирующей слабости и атрофии дистальной мускулатуры ног, угнетение сухожильных рефлексов (в первую очередь ахилловых), расстройства чувствительности по полипев- ритическрому типу, деформация стоп («стопы Фридрейха»), расстройства походки по типу «степпажа»; на поздней стадии может присоединяться слабость и атрофия дистальных отделов рук, деформация кистей [Dyck Р. et а!., 1993]. В основе болезни лежат дегенеративные изменения миелиновой оболочки или аксонов двигательных и чувствительных волокон периферических нервов и спинномозговых корешков. Заболеваниям из группы НМСН свойственен различный (как аутосомный, так и Х-сцепленный) характер наследования.
В соответствии с данными электрофизиологичес- ких и морфологических методов исследования можно четко дифференцировать 2 основных типа НМСН - НМСН I и НМСН II [Dyck Р. et al., 1993; Timmerman V. et al., 1998]. Тип I НМСН характеризуется, по данным электронейромиографии, значительным снижением скорости проведения по двигательным (менее 38 м/с) и чувствительным волокнам периферических нервов, а морфологически - сегментарной гипертрофической демие- линизацией нервов с формированием «луковичных головок» («onion bulbs»). Таким образом, НМСН I представляет собой демиелинизирующую форму полиневропатии (.миелинопатию). Напротив, для НМСН типа II характерно первичное поражение аксонов периферических нервов, при этом скорости проведения импульса по периферическим нервам в пределах нормы или умерен
но снижены, а на биопсии структура миелина остается сохранной (аксональная форма полиневропатии, или аксонопсття).
Помимо указанных выше двух основных типов моторно-сенсорных полиневропатий, иногда в литературе под рубрикой ЫМСН выделяют также ряд сравнительно редких синдромов, отличающихся от классического фенотипа Шарко-Мари-Тута теми или иными особенностями клинической картины [Dyck Р. el al., 1993; Harding А., 1995; Timmerman V. et al., 1998]:
а) HMCH III (синдром Дежерина-Сотта) - характеризуется дебютом симптомов на протяжении первых лет жизни, резко выраженной гипертрофией периферических нервов («гипертрофический неврит»), выраженным снижением скорости проведения импульса по двигательным волокнам (менее 10 м/с), ранней инвалидиза- цией и нередко наблюдаемым повышением уровня белка в ликворе;
б) HMCH IV (болезнь Рефсума) - самостоятельное заболевание, связанное с нарушением обмена фита- новой кислоты, при котором двигательная полипевро- патия сочетается с атаксией, ихтиозом и другими симптомами;
в) редкие формы 1ТМСН, характеризующиеся сочетанием преимущественно моторной невропатии с нижним спастическим иарапарезом (I IMC11V), атрофи ей зрительных нервов и глухотой (HMCH VI), пигментным ретинитом (HMCH VII);
г) врожденная гигюмиелинизирующая полиневропатия - характеризуется нарушением формирования миелииовой оболочки периферических нервов с рождения, значительным отставанием ребенка в двигательном развитии, резким снижением скорости проведения импульса по периферическим нервам.
3.1.3.1. Демиелинизирующие моторно-сенсорные невропатии (НМСН тип I)
Демиелинизирующие формы НМСН (НМСН I) являются гораздо более распространенными, чем аксональные. В абсолютном большинстве случаев заболевания из группы НМСН I наследуются по аутосомно-до- минантному типу. В настоящее время идентифицированы 4 гена аутосомно-доминантных НМСН I.
СМТ 1А. Основной ген, ответственный за развитие большинства случаев НМСН I, расположен в хромосомной области 17р11.2 и кодирует синтез структурного белка периферического миелина РМР22 {Peripheral Myelin Protein) [Lupski J. et al., 1991; Patel P. et al., 1992; Timmerman V. et al., 1992; Valentijn L. et al., 1992]. Данный хромосомный локус и соответствующая форма моторно-сенсорной невропатии получили обозначение СМТ типа 1А (аббревиатура от англ. Chare о l-Ма г i е— Tooth). Тип наследования заболевания - аутосомно-до- минантный. СМТ 1А имеет необычный молекулярный механизм, который определяется особенностями строения критической хромосомной области 17pl 1.2. Данный механизм схематично представлен на рис. 41.
Как видно на схеме, миелиновый ген РМР22 фланкируется высокогомологичными повторами ДНК, между которыми в мейозе может происходить неравный крос- синговер как результат спаривания ложно ориентированных друг против друга теломерного и центромерного повторов. В конечном счете образуются 2 различные мутантные хромосомы: одна из них содержит тандемную дупликацию области длиной 1,5 мб, другая - делецию той же области, причем в обоих случаях область хромосомной перестройки захватывает ген РМР22 [Chance Р. et al., 1994]. Таким образом, ген РМР22 является дозо- чувствительным'. гаметы с дупликацией РМР22 дают начало СМТ 1 А, тогда как делеция этого гена лежит в ос-
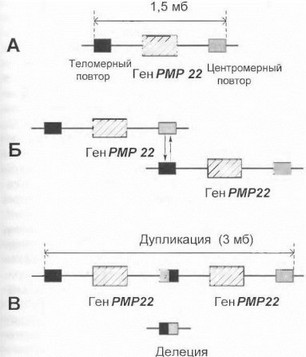
Рис. 41. Механизм неравного кроссинговера с участием гена РМР22
\ ( труктура кршической хромосомной области ] 7р 11.2. Ь. Неравный кроссинговер между двумя хроматидами (гомологичные тело- м( рный и центромерный повторы разных хроматнд спариваются мгж iy собой, приводя к неэквивалентному обмену хромосомными у час гками). В. Продукты неравного кроссинговера: показано обрати,шие двух мутантных хромосом с дупликацией и делецией ГМ1'22-содержащего хромосомного участка.
нове развития особой формы доминантного заболевания периферических нервов - наследственной невропатии с предрасположенностью к параличам от сдавления (см. ниже). Показано, что около 70-80% больных демиели- низирующими моторно-сенсорными невропатиями имеют указанную дупликацию в локусе 17р 11.2 [Wise С. et al., 1994], причем она часто возникает de novo в результате многократной реализации рассмотренного механизма неравного кроссинговера [Chance R, Fischbeck К., 1994]. В отдельных случаях заболевание обусловлено толковыми мутациями гена РМР22 [Roa В. et al., 1993; Nelis
Е. et al., 1994], что подтверждает этиологическую роль данного гена в развитии СМТ 1А. По-видимому, следствием указаиных толковых мутаций является повышение уровня экспрессии белка РМР22 - аналогично тому, что имеет место при дупликации гена. Предполагается, что изменение уровня экспрессии РМР22 может нарушать баланс между процессами пролиферации, остановки роста и дифференцировки шванновских клеток, приводя в конечном счете к гипертрофической демиелини- зации периферических нервов [Gabriel J. et al., 1997].
Другие молекулярные формы аутосомно-доминан- тных НМСН I. В более редких случаях доминантная моторно-сенсорная невропатия может быть связана с мутациями генов миелшювого белка Р0 на хромосоме Ц22-23 (СМТ типа IB) [Ilayasalca К. et al., 1993; Kulkens Т. et al., 1993] и коннексина-32 (Сх32) на хромосоме Xql3.1 (СМ1 типа IX) [Bergoffen J. et al., 1993] Форма СМТ 1В наследуется по аутосомно-доминаптно- му, а СМТ IX - по Х-сцепленному доминантному типу с ограниченной пенетран гностью у женщин. Белок Рп является важнейшим компонентом периферического ми елипа, рег\лирующим укладку слоев миелиной оболов ки, а конпексин-32 относится к белкам межклеточных контактов и формирует связи между соседними швап
новскими клетками (коннексиновые каналы) [Chance R, Fischbeck К., 1994]. К настоящему времени в данных генах описано большое число толковых мутаций (более 50 в гене Р0 и более 150 в гене Сх32), приводящих к нарушению структурно функциональной организации миелиновой оболочки периферических нервов и развитию СМГ 1В и СМТ IX [Nells Е. et al., 1999]. Клиническая картина этих форм НМСН практически неотличима от СМТ 1А. Недавно был идентифицирован еще один ген аутосомно-доминантной 11МСН типа I: он расположен на хромосоме 10q21.1 —22.1 и кодирует белок EGR2 (Early Growth Response), предположительно являющийся фактором транскрипции и контролирующий пролиферацию и миелинизацию шванновских клеток | Warner L. et al., 1998]. Мутации гена EGR2 являются чрезвычайно редкой причиной развития аутосомно-доминантной НМСН 1 (описано несколько толковых мутаций EGR2 в единичных семьях).
Синдром Дежерина-Сотта, врожденная гипоми- глинизирующая полиневропатия и синдром Русси-Леви.
I Интенсивные исследования генов РМР22, Р0 и EGR2 ползали, что данные гены ответственны не только за классические аутосомпо-доминаптные случаи НМСЕ1 типа I. но также за развитие описанных выше тяжелых (нередко врожденных) форм демиелинизирующих невропа- I w и - синдрома Дежерина-Сотта и врожденной гииомй- сипнизирующей полиневропатии [Nelis Е. et al., 1999].
1 [лыпые заболевания представляют собой экстремальные I и рианты фснотинического спек тра демиелинизирующих 111 пропатий: они обусловлены наиболее тяжелыми по lt; поим последствиям гетерозиготными толковыми мутациями de novo генов РМР22, Р0 и EGR2 или гомозиготными мутациями EGR2 и РМР22 (т.е. двойной дозой му Iиптного гена), в результате чего грубо нарушаются mi хамизмы формирования миелиновой оболочки периферических нервов [Chance Р., Fischbeck К., 1994; Haites N. et al., 1998; Parman Y. et al., 1999; Warner L. et ah, 1999]. Исходя из вышесказанного, методы ДНК-диагностики в указанных случаях синдрома Дежерина- Сотта и врожденной гипомиелинизирующей полиневропатии не отличаются от методов ДНК-диагностики, применяемых для всей группы НМСН I (см. ниже). Совсем недавно был открыт еще один генетический вариант синдрома Дежерина-Сотта с аутосомпо-рецессивиым типом наследования [Boerkoel С. et al., 2001]: он обусловлен мутациями в гене периаксина - белка цитоскелета, участвующего в реализации мембранно-белковых взаимодействий и стабилизации структуры зрелого миелина.
На протяжении многих десятилетий в литературе описывалось особое заболевание, известное под названием «синдром Русси-Леви». Для него характерно развитие типичной моторно-сенсорной невропатии с низкими скоростями проведения импульса по периферическим нервам, а также «полая стопа», постуральный тремор и легкие дискоординаторные симптомы [Dyck Р. et al., 1993; Harding А., 1995]. Многими авторами синдром Русси-Леви рассматривался как «стертая форма» болезни Фридрейха [Harding А., 1995]. Развитие электрофи- зиологических, морфологических и молекулярно-генетических методов показало, что синдром Русси-Леви не является самостоятельной нозологической формой, а представляет собой своеобразный фенотипический вариант НМСН I: данные электронейромиографии и биопсии нервов у больных с синдромом Русси-Леви идентичны таковым при НМСН I [Dyck Р. et al., 1993], а молекулярно-генетический анализ выявляет у этих больных мутации в генах Ро и РМР22 [Plante-Bordeneuve V. et al., 1999]. Таким образом, термин «синдром Русси- Леви» имеет лишь историческое значение и в настоящее время не применяется.
Наследственная невропатия с предрасположенностью к параличам от сдавления. Наследственная невропатия с предрасположенностью к параличам от сдавления (ННППС) представляет собой аутосомно-доминан- гное заболевание, характеризующееся развитием рецидивирующих демиелинизирующих моноиевропатий, обусловленных повышенной чувствительностью периферических нервов к сдавлению [Иллариошкин С.Н. и др., 1998; Gouider R. el al., 1995; Stogbauer F. et al., 2000]. У больных с молодого возраста отмечаются острые повторные эпизоды безболевых параличей периферических нервов, проявляющиеся парезами, парестезиями и расстройствами чувствительности в соответствующих зонах. Развитию параличей способствуют мелкие травмы и даже весьма незначительное и кратковременное сдавление нервов, например - после работы за письменным столом (повреждение локтевого нерва) или при сидении нога на ногу, на коленях, на корточках (паралич малоберцового нерва). В 10% всех случаев развития параличей наблюдается полное восстановление в течение первых 24 часов; более характерным является отсроченное восстановление (на протяжении ряда месяцев), в том числе с сохранением той или иной резидуальной симптоматики [Stogbauer F. et al., 2000]. По мере прогрессирования болезни постепенно могут развиваться симметричные или асимметричные амиотрофии в дистальных отделах конечностей, свисающая стопа, угнетение сухожильных рефлексов, «пятнистые» или диффузные расстройства чувствительности, что сближает клиническую картину ННППС с симптоматикой HMCFII. При элект- рофизиологическом исследовании у больных HI 1ППС отмечается снижение скорости проведения по двигательным и чувствительным волокнам периферических нервов, наиболее отчетливое в местах компрессии нервных стволов, а также удлинение дистальной латентности [Verhagen W. et al., 1993; Gouider R. et al., 1995}. Биопсия нервов выявляет характерные изменения миелина с формированием колбасовидных утолщений - так называемых «томакул» (отсюда синоним заболевания - «томакулярная невропатия»), а также сегментарную де- миелигшзацию [Verhagen W. et al., 1993; Siogbauer F. et al., 2000].
Как указывалось выше, молекулярной основой ННП11C является деления области длиной 1,5 мб (включая ген РМР22) как следствие неравного кроссинговера хромосомных участков в локусе 17р 11.2 (см. рис. 41) [Chance Р. et al., 1993; 1994]. Таким образом, ННППС и СМТ 1А обусловлены разнонаправленными нарушениями дозы гена РМР22 (деления или дупликация) в результате одного и того же мутационного события. Такой механизм предполагает, что распространенность НШ1ПС и СМТ 1А в популяции должна быть примерно одинаковой - около 1 на 3000 [Chance R, Fischbeck К., 1994]. Между тем истинная распространенность ННППС в прошлом явно недооценивалась, что обусловлено относительно доброкачественным течением заболевания и отсутствием выраженных клинических симптомов у большого числа носителей мутантного гена, нередко не обращающихся за медицинской помощью [Gouider R. et al., 1995].
В единичных случаях к развитию РШГ1ПС могут приводить также точковые мутации гена РМР22, приводящие (как и делеция гена) к снижению экспрессии РМР22 и дезорганизации периферического миелина [Stpgbauer F. et al., 2000].
Прямая ДНК-диагностика а^ тосомно-доминант- чых НМСН F Принимая во внимание тот факт, что около 80% всех случаев НМСН I обусловлены мутациями гена РМР22, обычно ДНК-диагностика НМСН I начинается с исследования именно этого гена и предполага
ет в первую очередь выявление основной мутации - дупликации области 1,5 мб в локусе 17р 11.2. Наиболее распространенным подходом для этого является исследование числа аллелей высокополиморфных (СА)п-маркеров, входящих в состав дуплицируемого участка [Cudrey С. etal., 1995]. В норме любой маркер имеет по два аллеля (по одному с каждой хромосомы), тогда как в случае дупликации на мутантной хромосоме 1,5 мб-участка общее число аллелей каждого маркера у больного СМТ 1А равно трем. Маркеры амплифицируются в ПЦР, после чего продукты амплификации разделяются с помощью электрофореза в полиакриламидном геле (рис. 42).
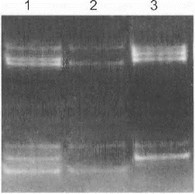
Рис. 42. ДНК-диагностика СМТ 1А с использованием полиморфных маркеров D17SI22 (верхняя часть теля) и I)] 7S921 (нижняя часть геля)
} (орожкн 1 и 3 - больные СМТ 1 А, дорожка 2 - больной с другой формой наследственной невропатии. У больных СМ Г 1А отмечается дупликация исследуемой хромосомной области (в дорожке I ви- |уализируются по 3 аллеля каждого и шаркеров, в дорожке 3 визу- лчп.знруются 3 аллеля маркера D17S122 и двойная доза нижнего л я 1еля маркера D17S921). Отдельные аллели 1йучасмых маркеров обозначены стрелками.
Дупликация 17р 11.2 диагностируется либо на основании регистрации трех различных ПЦР-продуктов (если все аллели имеют различную длину), либо на основании двойной интенсивности сигнала одной из полос (если два аллеля являются одинаковыми и «накладываются» друг на друга на электрофореграмме). Разновидностью указанного метода анализа полиморфных маркеров из области 17pl 1.2 является флюоресцентная гибридизация щ situ (FISH) с маркерными зондами, позволяющая регистрировать у больных СМТ 1А три пятна от гибриди- зованного флюоресцирующего маркера [Lupski J. et al., 1991]. Существует также ряд других, более сложных в техническом плане подходов для прямой регистрации дупликации критической хромосомной области 17р 11.2: а) блот-гибридизация по Саузерну с использованием различных зондов из дуплицируемой области; б) гель-элек- трофорез в пульсирующем поле, позволяющий регистрировать изменение подвижности сверхкрупных (дуплицированных) фрагментов ДНК, полученных после рестрикции геномной ДНК и гибридизованных с соответствующими зондами; в) обнаружение (на основе рестрикции и последующей гибридизации) рекомбинантного участка в локусе 17р11.2, послужившего источником дупликации [Patel Р. et al., 1992; Raeymaekers Р. et al., 1992; Timmerman V. et al., 1997].
При отсутствии данных за дупликацию в области 17р 11.2 проводится последовательный поиск толковых мутаций в генах РМР22, Р0, Сх32 и ERG2. Порядок исследования генов может варьироваться в зависимости от относительной частоты встречаемости мутаций в них в различных популяциях, а также в зависимости от особенностей генеалогии конкретной родословной: например, при обследовании семьи с подозрением на Х-сцеп- ленный доминантный тин наследования НМСН I в пер
вую очередь должен исследоваться ген Сх32. Для поиска толковых му гаций обычно используются стандартные процедуры скрининга отдельных экзонов (SSCP-анализ, гетеродуплексный анализ и др.) с последующим прямым секвенированием предположительно муз аЗнтн ых образцов ДНК либо тотальное секвенирование всей кодирующей области гена [Nelis Е. et al., 1999].
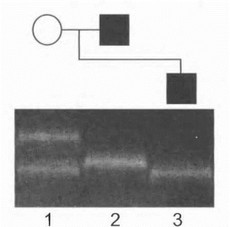
Рис. 43. ДН К-диагностика наследственной невропатии с предрасположенностью к параличам от сдавления с использованием полиморфного маркера D17S921 У больных отца и сына (дорожки 2 и 3) отмечается «потеря i етерозиготностн» и наличие лишь одного маркерного аллеля. Вследствие делецнн изучаемой хромосомной области у сына на- о.шодается видимое «отсутствие» отцовского аллеля (сын унаследовал от больного отца мутантную хромосому, не дающую i (родукта амплификации).
Для диагностики делеции в области 17р11.2 у бlt; gt; ньных НБ ППС используются те же методы, что и для
выявления дупликации при СМТ 1А. При этом анализ высокополиморфных маркеров (С А) позволяет диагностировать феномен «потери гетерозиготпости» у больного родителя и отсутствие родительского аллеля у больного ребенка (рис. 43). Диагностика делении возможна также на основании прямого обнаружения половинной дозы гена РМР22 (например, при блот-гибридизации ге- номной/амплифицированной ДНК или количественном определении ПЦР-продукта участка гена с использованием автоматического секвенатора). Нейрогенетическим отделением НИИ неврологии РАМН проведен молекулярно-генетический анализ большой серии рецидивирующих невоспалительных невропатий на основе исследования гена РМР22. Полученные нами результаты свидетельствуют о том, что дайеция РМР22 обусловливает около 30% случаев рецидивирующих невропатий различной локализации и более половины семейных форм данных заболеваний (в том числе при семейной невропатии лицевого нерва). Принимая во внимание сложность дифференциальной диагностики ННППС с другими формами наследственных невропатий, а также несомненно существующую недооценку распространенности ННППС, можно сделать вывод о том, что прямая ДНК-диагностика является ключевым методом исследования и позволяет в абсолютном большинстве случаев безошибочно диагностировать ННППС, не прибегая к необходимости проведения такой инвазивной процедуры как биопсия нерва. ДНК-диагностика должна применяться не только у больных, но и у их клинически здоровых родствен ников из группы риска, поскольку заболевание может иметь стертое и асимптомное течение. Больным со своевременно диагностированной ННППС необходимо рекомендовать соответствующую коррекцию стиля жизни физических нагрузок и профессиональной ориентации что имеет решающее значение для профилактики необратимых и инвалидизирующих осложнений данного заболевания. ДНК-диагностика ННЛПС позволяет избежать необоснованного направления больного к хирургу, поскольку оперативное лечение, являющееся в ряде случаев наиболее эффективным и радикальным методом терапии обычных туннельных компрессионных синдромов, при ННЛПС неэффективно и лишено смысла.
Аутосомно-рецессивиые формы НМСН 1. Деми- елинизирующие моторно-сенсорные невропатии могут наследоваться и по аутосомно-рецессивному типу (СМТ 4). Такие заболевания представляют собой исключительную редкость и описаны в единичных семьях [Harding А., 1995]. При подозрении на аутосомно-рецес- сивную форму ПМСН 1 необходимо тщательное клиническое и электрофизиологическое обследование родителей пробанда с целью исключения у них субклиничсски протекающей невропатии, т.е. для исключения аутосом- но-доминатного наследования. На сегодняшний день известны 7 самостоятельных генетических вариантов ауто- сомно-рецессивных демиелинизирующих моторно-сенсорных невропатий - они сцеплены с хромосомами 8q 13-21 (СМТ 4А) [Ben Othmane К. et al., 1993 (а)], 11 q23 (СМТ 4B1) [Bolino A. et al., 1996], 11 p 15 (CMT 4В2) [Ben Othmane K. et al., 1999], 5q23-33 (СМТ 4C) 11 ,e Guern E. ex al., 1996], 8q24.3 (CMT 4D) [Kalaydjicva L. cl al, 1996], 10q21.1-22.1 (CMT4E) [Warner E. etal., 1998] и 19ql3.1-13.3 (CMT 4F) [Dclague V. et al, 2000]. Bee сказанные формы аутосомно-рецессивных демислиии- шрующих моторно-сенсорных невропатий описаны в семьях с кровнородственными браками. Общими особенностями клинической картины являются более тяже- щgt;е течение и частое сочетание с глухотой и атрофией '.ри гельных нервов.
Гены 3 форм аутосомно-рецессивных демиелини- зирующих моторно-сенсорных невропатий к настоящему времени идентифицированы. Форма СМТ 4В1 обусловлена мутациями в гене тирозин-фосфатазы (MTMR2), имеющей отношение к белку аксонального цитоскелета миотубулярину [Bolino A. et al., 2000]; детальные механизмы повреждения асконов периферических нервов при СМТ 4В1 не установлены. Форма СМТ 4Е представляет собой тяжелую врожденную гипомиелинизирующую полиневропатию, обусловленную гомозиготностью по мутациям в гене EGR2 [Warner Е. et al., 1998]; как указывалось выше, гетерозиготные мутации в этом же гене являются причиной развития более мягкой аутосомно- доминантной формы демиелинизирующей моторно-сенсорной невропатии. Наконец, в основе развития СМТ 4D лежат мутации в гене сигнального белка NDRG1, регулирующего процессы роста и дифференцировки шван- новских клеток и взаимодействие между шваниовскими клетками и аксоном, а также участвующего в поддержании Целостности структуры зрелого аксона [Kalaydjieva L. et al., 2000]. Интересно отметить, что ген еще одной формы - СМТ 4F - расположен в том же локусе на хромосоме 19ql 3, что и ген, кодирующий белок цитоскелета периаксин, мутации в котором вызывают аутосомно-рецессивный вариант синдрома Дежерина- Сотта (см. выше). Весьма вероятно, таким образом, что СМТ 4F и указанный вариант синдрома Дежерина-Сот- та являются аллельными заболеваниями.
В связи с тем, что четкие фенотипические различия между отдельными формами аутосомно-рецессивных демиелинизирующих моторно-сенсорных невропатий (за исключение СМТ 4Е) отсутствуют, прямая ДНК- диагностика данных заболеваний возможна только после обнаружения достоверного сцепления с соответствующими хромосомами. При развитии тяжелого аутосом- но-редессивного варианта гипомиелинизирующей полиневропатии, прояв тающейся с момента рождения ребенка, можно диагностировать форму СМТ 4Е и предпринять исследование гена EGR2, которое проводится стандартными методами мутационного скрининга. В остальных случаях при обследовании больших информативных родословных возможно проведение анализа генетического сцепления с маркерами из соответствующих локусов и в случае подтверждения высокого Лод-балла- проведение косвенной ДНК-диагностики у членов семьи из группы риска.
- Аксональные моторно-сенсорные невропатии (НМСН тип II)
НМСН типа II являются относительно редкими заболеваниями (4-12 случаев на 100 000) [De Jonghe Р. ct al., 1998]. Большинство заболеваний из группы НМСН 11 наследуются по аутосомно-доминантному типу, описаны также отдельные семьи с аутосомно-рецессивным и Х-сцепленным рецессивным наследованием | Harding А., 1995]. По сравнению с НМСН I, для IIMCH II характерен более поздний возраст начала бо- |[сзни (в среднем на 10 лет позже), меньшее вовлечение мелких мышц кисти и меньшая степень угнетения сухожильных рефлексов; при отдельных молекулярных вариантах НМСН II может выявляться также ряд дополнительных симптомов, не свойственных демиелинизи- рующим формам НМСН [Dyck Р. et al., 1993; Chance Р, I ischbeck К., 1994].
Молекулярная генетика аксональных форм мотор- I ю-сенсорных невропатий остается малоизученной. Име- || мциеся данные свидетельствуют в пользу несомненной 1гпстической гетерогенности НМСН II: на различных ромосомах картированы как минимум 7 самостоятель- Н ы х локусов НМСН II. Аутосомно-доминантные формы НМСН II сцеплены с хромосомами 1 р35—36 (локус
СМТ2А), 3ql3—22 (СМТ 2В), 7р14 (СМТ 2D) и 7qll- 21 (СМТ 2F) [Ben Othmane К et al., 1993 (b); Kwon J. et al., 1995; Ionasescu V. etal., 1996; Ismailov S. etal., 2001]. Характерной особенностью формы СМТ 2В является выраженный сенсорный компонент и развитие трофических язв нижних конечностей, а СМТ 2D - более отчетливое и раннее вовлечение рук. Совсем недавно группой российских ученых из Медико-генетического научного центра РАМН был идентифицирован еще один генетический вариант доминантной аксональной моторно-сенсорной невропатии (СМТ 2Е), связанный с мутацией гена одного из белков аксонального цитоскелета - белка легкого нейрофиламента (ген расположен на хромосоме 8р21) [Mersiyanova I. et al., 2000]. Известны два локуса аутосомно-рецессивной формы НМСНII: они картированы на хромосомах lq21.2-21.3 [Bouhouche A. et al., 1999] и 19ql3.3 [Leal A. et al., 2001]. Наконец, X- сцепленная рецессивная форма НМСН II, ассоциированная с глухотой и умственной отсталостью (синдром Cowchock), связана с хромосомным локусом Xq24-26 [Priest J. et al., 1995]. В отличие от перечисленных дистальных вариантов НМСН II (фенотип Шарко-Мари- Тута), Takashima Н. с соавторами описали особую форму аксональной 11МСН с преимущественно проксимальным типом вовлечения скелетной мускулатуры, ген дан ного аутосомно-доминантного заболевания был картирован на хромосоме 3q 13.1 [Takashima Н. et al., 19991 Для всех генетических вариантов аксональных НМСЗI при наличии достаточно больших родословных возможно исследование высокополиморфных маркеров из ук:1 занных хромосомных локусов с целью анализа генетп ческого сцепления; при получении достоверных данны n в пользу сцепления с определенным локусом может про водиться косвенная ДНК-диагностика у родственникиi. из группы риска. В 8р21-сцепленных семьях с доминап тной формой аксональной НМСН (СМТ 2Е) возможна прямая ДНК-диагностика болезни на основе обнаружения мутации в гене легкого нейрофиламента.
Описаны казуистические случаи, когда у больных с фенотипом НМСН II (т.е. при нормальных или субнормальных скоростях проведения импульса по периферическим нервам) при ДНК-анализе выявлялись мутации в генах Сх32 и Р0 —т.е. молекулярные дефекты, обычно' свойственные демиелинизирующим формам наследственных невропатий [De Jonghe Р. et a.L, 1998]. Мутации в указанных генах могут выявляться также у больных, имеющих при электронейромиографическом исследовании «промежуточные» значения скоростей проведения импульса (между 30 и 40 м/с). Таким образом, данные гены должны становиться объектами анализа в I ех редких случаях, когда точное отнесение наследственной невропатии к демиелинизирующим или аксональным формам на основании клинических, электрофизио- ногических и гистологических данных затруднено. По мнению Timmerman V. et al. (19966), мутационный скрининг гена Сх32 целесообразно проводить при НМСН 11, когда в обследуемой родословной не зарегистрировано случаев передачи болезни от отца сыну (т.е. когда не ис- к мочен Х-сцеплегшый тип наследования, характерный мя СМТ IX).