
Среди всех спинал ьных амиотрофий заболевания с ранним началом и преимущественным вовлечением проксимальной мускулатуры являются наиболее распро- страненными и изученными формами (1:10 000) [Emery А., 1991; International SMA Consortium, 1992]. Тип наследования в абсолютном большинстве случаев - ауто- сомно-рецессивный. В соответствии с особенностями течения проксимальные аутосомно-рецессивные спинальные амиотрофии подразделяются на три основных клинических варианта: 1) острая инфантильная форма Верднига-Гофмана (1-й тип спинальной амиотрофии) - характеризуется развитием тяжелой генерализованной мышечной слабости и гипотонии на протяжении первых 3 месяцев жизни и наступлением летального исхода вследствие дыхательных нарушений на 1 -2 году жизни ребенка; 2) «промежуточная» или хроническая инфантильная форма (2-й тип), при которой ребенок заболевает в раннем детстве, может научиться самостоятельно сидеть, однако обычно не способен стоять или ходить без поддержки; 3) хроническая ювенильная форма, или болезнь Кугельберга-Веландер (3-й тип) - характеризуется началом болезни па 1 -м или 2-м десятилетии жизни и более медленным течением на протяжении десятилетий [International SMA Consortium, 1992].
Все три формы аутосомно-рецессивной спинальной амиотрофии являются аллельными заболеваниями, обусловленными мутациями одного гена, локализованного на длинном плече 5-й хромосомы [Gilliam Т. et al., 1990; Melki J. et al., 1990]. Данный ген, идентифицированный в 1995 г. и состоящий из 8 экзонов, получил обозначение SMN (аббревиатура от англ. Survival Motor Neuron - «ген выживаемости мотонейрона») | Lefebvre S. et al., 1995]. Ген SMN представлен в области 5ql3 двумя копиями - теломерной (SMNT), расположенной дистально ближе к теломере хромосомы, и центромерной (SMNC), расположенной более проксимально [Lefebvre S. etal., 1995]. Высокая нуклеотидная гомология между SMN1 и SMNC способствует нарушению нормальных механизмов кроссинговера и выраженной генетической нестабильности критического участка хромосомы 5ql3, результатом чего является образование I амет, в которых ген SMN1 утрачен или замещен его центромерной копией SMNC [Hahnen Е. et al., 1996; ( ampbell L. et al., 1997; Lefebvre S. et al., 1998]. В целом около 98,6% больных проксимальной спинальной ами- lt; п рофией имеют гомозиготную делецию всего гена SMN1 иибо его части [Cobben J. et al., 1995; Lefebvre S. et al., 1995; Rodrigues N. et al., 1995]; в то же время центромерные копии гена обычно остаются интактными либо обнаруживаются в увеличенном числе копий [Taylor .1. ¦I al., 1998; Vital' Т. et al., 1999]. В единичных случаях проксимальной спинальной амиотрофии показано наличие патогенетически значимых точковых мутаций в гене '!MNT [BussagliaE. et al., 1995; Lefebvre S. et al., 1995]. Все эти данные свидетельствуют о непосредственном иоклечении теломерной копии гена SMN (SIVfNP) в па- I oi снез аутосомно-рецессивной проксимальной спинальной амиотрофии Поскольку для различных по тяжести вариантов спинальной амиотрофии (1-й, 2-й или 3-й тип) характерен один и тот же первичный молекулярный дефект - делеция гена SMNT, имеются дополнительные генетические факторы, модифицирующие характер клинических проявлений болезни. К таким факторам относятся протяженность делеции и вовлечение близлежащего гена белка-ингибитора апоптоза (N AIP), число сохранных копий гена SMNC и др. [Lefebvre S. et al., 1995; Velasco E. et al., 1996; Vitali T. et al., 1999]. Показано, в частности, что у больных с наиболее «мягким» фенотипом - болезнью Кугельберга-Веландер - число копий гена SMNC составляет в среднем 4, тогда как при тяжелой форме (болезнь Верднига-Гофмана) число копий SMNC обычно не превышает 1-2 [Campbell L. et al., 1997; Taylor J. et al., 1998; Vitali T. et al., 1999]. В связи с этим предполагается, что экспрессия гена SMNC имеет функциональное значение и в определенной степени может компенсировать (хотя бы частично) отсутствие у больных «основного» гена SMNT
Исследование гена SMN открыло возможность проведения прямой ДНК-диагностики аутосомно-рецес- сивной спинальной амиотрофии детского возраста. Основной технической проблемой при этом является необходимость дифференцировать теломерную (значимую) копию SMN от гомологичной центромерной копии. Нуклеотидные последовательности теломерной и ценгромер- ной копий гена SMN практически идентичны, за исключением 7-го и 8-го экзонов: по каждому из этих экзонов SMNT отличается от SMNC на один нуклеотид [Lefebvre S. et al., 1995]. Указанные различия нуклеотид ¦ ного состава SMNT и SMNC позволяют дифференциро вать эти гены с помощью сравнительно простого мето ¦ да, предложенного van der Steege G. с соавт. в 1995 году.
ДНК- диагностика наследспвенных болезней... 197
Метод основан на использовании рестрикционных эндонуклеаз, имеющих сайты распознавания в указанных вариабельных участках 7-го и 8-го экзонов SMN. Данные ферменты расщепляют амплифицированные экзоны SMNC на более короткие фрагменты, тогда как для тех же экзонов SMN'1 рестрикция невозможна (рис. 44).
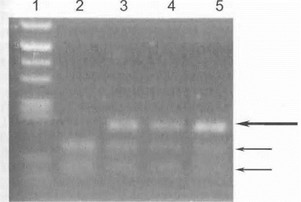
1*ис. 44. Прямая ДНК-Дйагностпкааутосомно-рецееоивной проксимальной спинальной амиотрофии детского возраста 11родукты амплификации 8-го тжзона гена SMN обработаны рест- I чпсгазой Dde I. Длинной стрелкой указан продукт амплификации iniaSMN1, короткими стрелками -продукт амплификации гена 4MNC, разреза! гный реетрнктазой на 2 фрагмента. Дорожка I - маркер, дорожка 2 - больной а шпальной амиотрофней Кугельбсрга- I(еландер (гомозиготная деления гена SM N1), дорожки 3,4- больные е другими молекуляриыми формами спинальных амиотрофии (IICT гомозиготной делении гена SMN1), дорожка 5 - контроль.
Таким образом, отсутствие SM№ -специфичных фраг ментов ДНК позволяет установить гомозиготную
делецию гена SMNth гем самым диагностировать ауто- сомно-рецессивную проксимальную спинальную амиот- рофию (формы Верднига-Гофмана, Кугельберга-Велагг- дер или «промежуточный» вариант болезни).
Как указывалось выше, у небольшого числа больных (lt;2%) на одной из мутантных хромосом может иметь место не полная делеция гена SMNT, а толковая мутация в нем, в связи с чем ПЦР-продукт данного аллеля будет визуализироваться на электрофореграмме. В этих случаях для Д1 [К-диагностики болезни необходимо определение дозы гена SMNT, т.е. доказательство наличия у больного одной копии SMNT по сравнению с двумя копиями данного гена в контрольных образцах. Для анализа дозы гена требуется постановка реакции в особых стандартизированных условиях (количественная ПЦР): отсутствие одной копии SMNT может быть определено либо визуально - на основании вдвое меньшей интенсивности окрашивания соответствующего фрагмента ДНК по отношению к норме, либо с помощью компьютерного анализа относительной площади флюоресценции пиков ДНК. Для этой же цели может использоваться денситометрия радиоавтографа, полученного при проведении блот-гибридизации. С практической точки зрения, выявление половинной дозы гена SMNT (т.е. его делеции в гетерозиготном состоянии) позволяет, при наличии соответствующей клинической картины, с высокой степенью вероятности склониться к диагнозу ауто- сомно-рецессивной 5ql3-c4emieHHofi спинальной ами- отрофии. Однако достоверная диагностика в этом случае возможна только при условии идентификации толковой мутации во втором аллеле гена.
Наш собственный 4-летний опыт проведения прямой ДНК-диагностики данных заболеваний в семьях из европейской и центральной части России показал, что при анализе строго отобранной серии классических форм спинальной амиотрофии более чем в 90% случаев выявляется гомозиготная делеция 7-го и 8-го экзонов гена SMNT. Это соответ ствует данным большинства авторов и позволяет заключить, что в нашей стране данный тип мутации также является ведущей причиной развития аутосомно-рецессивной спинальной амиотрофии детского возраста. Таким образом, сравнительно простая ДНК-диагностика спинальной амиотрофии возможна в абсолютном большинстве случаев данного заболевания. Следует отметить, однако, что в отдельных популяциях (особенно характеризующихся высоким уровнем инбридинга и «эффектом основателя») соотношение основных типов мутаций может иметь существенные особенности. Так, нами совместно с капд. мед. наук Г.Х. Багыевой в 1998-1999 гг. были обследованы 8 семей с аутосомно- рецессивной спинальной амиотрофиея из Ахалского ве- лаята Туркменистана: гомозиготная делеция 7-го и 8-го экзонов гена SMNT была выявлена лишь в 2 семьях, что может свидетельствовать о накоплении других мутаций SMNT либо о генетической гетерогенности данного заболевания в туркменской популяции.
Диагностика гетерозиготного носительства делении гена SMN1 у родителей больных спинальной ами- отрофией основывается на описанных выше методах определения дозы гена. В случае выявления у обоих родителей носительства делении в гетерозиготном состоянии можно сделать вывод о том, что каждая из мутантных хромосом унаследована пробандом от родителей, а не возникла de novo. В такой ситуации риск повторного возникновения заболевания в семье для каждого из сиб- сов равен 25%, что требует подтверждения с помощью прямой ДНК-диагностики (в том числе возможно проведение пренатальной ДНК-диагпостики с целью профилактики рождения больного ребенка).
- Спинально-бульбарная амиотрофия Кеннеди (болезнь Кеннеди)
Спинально-бульбарная амиотрофия Кеннеди - редкое заболевание, характеризующееся Х-сцепленным рецессивным типом наследования и проявляющееся у мужнин в относительно позднем возрасте (обычно после 40 лет). Типичная клиническая картина включает медленно прогрессирующую мышечную слабость, амиотро- фии и фасцикуляции проксимальных отделов конечностей, бульбарные симптомы денервационного характера (дизартрия, дисфагия, фибрилляции языка), а также характерные эндокринные расстройства (гинекомастия, тестикулярная атрофия) [Kennedy W. et al., 1968]. На поздней стадии может вовлекаться проксимальная мускулатура ног.
Заболевание обусловлено повреждением гена андрогенного рецептора, расположенного в локусе Xql 1.2- 12 [La Spada A. et al., 1991]. У всех больных амиотрофи- ей Кеннеди имеет место экспансия тандемных тринук- леотидных повторов CAG в 1-м экзоне гена: в норме число копий CAG-повторов составляет 9-36, тогда как больные амиотрофией Кеннеди имеют увеличенное число тандемных повторов - от 38 до 72 [La Spada A. et al., 1991; Igarashi S. et al., 1992; Amato A. et al., 1993]. Такой характер мутации на белковом уровне проявляется патологическим удлинением соответствующего нолиглута- минового участка белка, что лишь в небольшой степени влияет на нормальную функцию андрогенного рецептора (у больных отмечается лишь умеренное снижение чувствительности к действию андрогенов). Как и при других «полиглутаминовых» болезнях, поражение ЦНС при болезни Кеннеди связывается с тем, что мутантный бе- *ок приобретает новые цитотоксические свойства и способствует формированию патологических внутриядер ных включений [Mhatre A. etal., 1993; HousmanD., 1995.
Li М. et al., 1998]. При этом с увеличением числа CAG- повторов и длины полиглутаминового участка заболевание характеризуется более тяжелым течением и более ранним началом [Igarashi S. et al., 1992; La Spada A. et al., 1992]. Интересно отметить, что точковые мутации в данном гене, приводящие к инактивации андрогенного рецептора, сопровождаются развитием совершенно другого заболевания -так называемого синдрома тестикулярной феминизации [Gottlieb В. et al., 1998]. Таким образом, различные по своей сущности мутации в гене андрогенного рецептора, по-разному влияющие на функцию данного белка, лежат в основе принципиально различ- ных форм патологии.
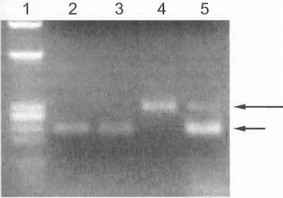
1*ис 45. Прямая ДНК-диагност.ика спинально-бульбарной i ч I ютрофии Кеннеди
)(igt;рожка 1 - маркер, дорожки 2,3 - контроль, дорожка 4 - больной • I шпально-бульбарной а'миотрофией Кеннеди, дорожка 5 - мать i к пп.ного (гетерозиготная носительница мутации). Длинной стрелкой указа! мутантный аллель (экспансия CAG-повторов гена андрогенною рецептора), короткой ст редкой- нормальный аллели.
Прямая ДНК-диагностика болезни Кеннеди является относительно несложной и основана на ПЦР-ам- плификации фрагмента 1-го экзона гена, содержащего тринуклеотидный участок. У больных мужчин мутантный аллель (продукт единственной Х-хромосомы) четко определяется благодаря более медленной электрофоретической подвижности, что является следствием увеличенного числа тринуклеотидных CAG-повторов (рис. 45, дорожка 4). У женщин-носительниц на электрофо- реграмме визуализируются нормальный и мутантный аллели (рис. 45, дорожка 5), что позволяет достоверно диагностировать наличие мутации в гетерозиготном состоянии. В отягощенных семьях возможно проведение ранней пресимптоматической ДНК-диагностики болезни у лиц мужского пола, а также пренатальной ДНК- диагпостики.
- Семейный боковой амиотрофический склероз
Боковой амиотрофический склероз - тяжелое дегенеративное заболевание ЦНС позднего возраста (средний возраст начала составляет около 55 лет). Оно характеризуется прогрессирующей гибелью центральных и периферических мотонейронов с развитием парезов скелетной и бульбарной мускулатуры, амиотрофий, фасци- куляций и пирамидной спастичности; летальный исход (главным образом, вследствие дыхательных нарушений) наступает в среднем через несколько лет от момента появления первых симптомов болезни [Leigh R, Ray- Chaudhuri К., 1994]. Распространенность заболевания составляет 4-6 случаев на 100 000 населения. В большинстве случаев боковой амиотрофический склероз представляет собой спорадическое заболевание мульти- факториальной этиологии. У 5-10% больных, однако может выявляться положительный семейный анамнез,
свидетельствующий о моногенном аутосомно-доминан- тном или (гораздо реже) аутосомно-рецессивном наследовании заболевания среди родственников [Siddique Т, Deng Н., 1996]. Клиническая картина семейных и спорадических форм бокового амиотрофического склероза в большинстве случаев практически идентична (некоторые исключения рассмотрены ниже), однако в целом для семейных форм заболевания характерен более ранний дебют симтомов.
Приблизительно у 20% больных с аутосомно- доминантной формой бокового амиотрофического склероза заболевание обусловлено мутациями гена цитозольного фермента Cu/Zn супероксиддисмутазы (SOD1) на хромосоме 21q22 fRosen D. et al., 1993]. К настоящему времени известно свыше 50 мутаций, распределенных по всем экзонам SOD1 (они в основном представлены толковыми мутациями, реже вставками и делециями) [Siddique Т., Deng Н., 1996; Rowland L., 1998]. Некоторые из мутаций описаны лишь в отдельных семьях, другие являются мажорными в определенных популяциях. Так, мутация Ala4Val обусловливает половину всех случаев данной молекулярной формы семейного бокового амиотрофического склероза в Северной Америке [Cudkowicz М. et al., 1997]. Мутации в гене SOD1 обнаруживаются также у 2-5% больных со спорадической формой бокового амиотрофического склероза [Jones С. et al., 1995; Andersen R et al., 1997]. Предполагается, что в этих случаях имеет место «ложно-отрицательный семейный анамнез» (ранняя смерть или поздняя манифестация болезни у одного из родителей) либо неполная пенетрантность мутантного гена [Rowland L., 1998]. Интересно отметить, что мутация Asp90Ala в ряде случаев приводит к развитию болезни лишь будучи в гомо- шготном состоянии и, таким образом, может являться причиной аутосомно-рецессивной формы бокового амиотрофического склероза [Andersen Р. et al., 1997].
В казуистических случаях отдельные мутации SOD1 могут быть ассоциированы с весьма атипичными вариантами болезни - например, характеризующимися длительным течением свыше 20 лет [Aoki М. et al., 1994] либо отсутствием вовлечения верхнего мотонейрона [Cudkowicz М. et al., 1997; Rowland L., 1998]. В связи с этим для семейных случаев обсуждается возможность пересмотра классических клинических критериев бокового амиотрофического склероза (так называемых «критериев Эль-Эскориаль»), По мнению Cudkowicz М. et al. (1997), для достоверной диагностики семейных случаев болезни может быть достаточным наличие следующих двух признаков: 1) симптоматика поражения периферического мотонейрона в трех конечностях; 2) обнаружение мутации в гене SOD1. Таким образом, мутационный скрининг SOD 1 может являться ценной диагностической процедурой в семейных (особенно атипично потекающих) случаях бокового амиотрофического склероза. В семьях с идентифицированной мутацией возможно проведение прямой ДНК-диагностики у лиц из группы риска. В целом, однако, практическое значение ДНК- диагностики при данном заболевании остается на сегодня сравнительно ограниченным, поскольку вероятность выявления мутации невелика: как указано выше, в 80% аутосомно-доминантных случаев заболевание не связано с мутациями в гене SOD1.
В отличие от классической фатальной формы бокового амиотрофического склероза с поздним началом в литературе описаны редкие семьи так называемого ювенильного бокового амиотрофического склероза. Для него характерно начало болезни чаще всего на 2-м десятилетии жизни, вариабельное соотношение выраженности поражения центрального и периферического мотонейронов, а также весьма медленное прогрессирование, в некоторых случаях даже не влияющее на естественную продолжительность жизни [Siddique Т., Deng Н., 1996]. Ювенильный боковой амиотрофический склероз может наследоваться по аутосомно-доминантному и аутосом- но-рецессивному типу. Ген аутосомно-доминантной формы картирован на хромосоме 9q34 [Chance Р. et al., 1998]. Аутосомно-рецессивпая форма является генетически гетерогенной: один локус расположен на хромосоме 2q33 (тунисский вариант болезни), другой (более частый вариант болезни, обнаруживаемый в Северной Африке и Европе) - на хромосоме 15ql5.1-22.1 [Ilentati A. et al., 1994а; 1998]. Первичные молекулярные дефекты, ответственные за развитие ювенильного бокового амиотрофического склероза, до настоящего времени не установлены. В информативных семьях возможно проведение косвенной ДНК-диагностики на основе анализа сцепления с генетическими маркерами хромосом 2q, 9q и 15q.