
Детальный анализ клинического полиморфизма торсионной дистонии позволил Е.Д. Марковой в 1975 году подразделить это заболевание на две основные формы: ршидную и гиперкинетическую. Ригидная форма торсионной дистонии начинается обычно в детском или юношеском возрасте и характеризуется постоянным повышением мышечного тонуса по экстрапирамидному типу, формированием фиксированных патологических поз, развитием контрактур на поздней стадии болезни [Ткачев Р.А., Маркова Е.Д. и др., 1972; Маркова Е.Д., 1975; 1983]. Аналогичная форма торсионной дистонии была описана японскими исследователями под названием «наследственная прогрессирующая дистония» («hereditary progressive dystonia») [SegawaM. etal., 1972; 1976]. Segawa M. и соавторы отметили, что для данной клинической формы дистонии характерным является наличие дневных флюктуаций в моторике больных - ухудшение состояния к вечеру и улучшение двигательных функций утром или после дневного сна. Заболевание чаще наблюдается у женщин [Segawa М. et al., 1976; Markova Е., Ivanova-Smolenskaya I., 1995]. Еиперкинети- ческая форма торсионной дистонии, которая обычно начинается на 1-м десятилетии жизни, характеризуется развитием вычурных «скручивающих» генерализован ных гиперкинезов конечностей, туловища и шеи, резко усиливающихся при ходьбе и приводящих к тяжелой инвалидизации больного [Маркова Е.Д., 1975; 1989]. В западной литературе для обозначения данного заболевания чаще используются терминш «генерализованная дистония с ранним началом» («early-onset generelized dystonia») либо lt;«идиопатическая торсионная дистония» («idiopathic torsion dystonia») [ZiberN. etal., 1984; Fahn S., 1988; Jankovic J., Fahn S., 1998]. Гиперкинетическая форма торсионной дистонии особенно часто встречается в этнической группе евреев ашкенази - около 40-50 случаев на 100 000 [Ziber N. et al., 1984].
В основе фенотипических различий двух данных форм торсионной дистонии лежат разнонаправленные изменения метаболизма основных нейротрансмиттеров в головном мозге: при ригидной форме выявлено снижение центральной дофаминергической и серотопинер- гической трансмиссии с одновременным повышением активности холинергической системы, тогда как для ги- перкинетической формы характерны обратные соотношения [Бархатова В.П., 1988; LeWitte Р., 1993]. Это явилось теоретической предпосылкой для назначения больным с ригидной формой торсионной дистонии заместительной терапии препаратами Ь-дцфа. Такое лечение было предложено в начале 70-х годов и выявило «драматический» эффект малых доз пренарата, сохраняющийся па протяжении многих лет при отсутствии серьезных побочных эффектов [Ткачев Р.А. и др., 1972; 1974; Segawa М. et al., 1976]. Результаты лечения дали основание Nygaard Т. и соавторам в 1988 году предложить для обозначения данной клинической формы торсионной 'щетонии термин «дофа-зависимая» или «дофа-чувстви- щдьная дистония» («dopa-responsive dystonia»). Напро- шв, при гиперкинетической форме торсионной дистонии назначение L-дофа не вызывает улучшения состояния и даже приводит к усилению гиперкинезов («дофа- независимая дистония») [Маркова Е.Д., 1975; 1989; Иванова-Смоленская И.А. и др., 1998 (а)].
Дофа-зависимая дистония. В 1993 году Т. Nygaard с соавторами картировали ген аутосомно-доминантной дофа-зависимой дистонии на длинном плече 14-й хромосомы [Nygaard Т. et al., 1993]. Вскоре после этого локализация мутантного гена на хромосоме 14q была подтверждена японскими исследователями в семьях с «наследственной прогрессирующей дистонией» [Tanaka Н. et al., 1995], что позволило на молекулярном уровне установить нозологическую идентичность понятий «наследственная прогрессирующая дистония» и «дофа-зависимая дистония». Выделенный ген кодирует синтез одного из ключевых ферментов дофаминового обмена - ГТФ-циклогидролазу 1 (ГЦГ-1) [IchinoseH. etal., 1994]. В свете современных представлений патогенез аутосомно-доминантной дофа-зависимой дистонии схематично показан на рис. 51. Фермент ГЦГ-1 регулирует превращение ГТФ в дигидронеоптерин-трифосфат; последний, в свою очередь, является субстратом для образования тетрагидробиоптерина (ВН4) - кофактора тирозин-гид- роксилазы, ответственной за синтез дофамина из тирозина. При наличии мутаций в гене ГЦГ-1 нарушается функция данного фермента и весь последующий биохимический каскад, что приводит к недостаточности синтеза дофамина в нейронах черной субстанции [Ozelius Г., Breakefield X., 1994]. Синтез дофамина может быть эффективно восстановлен при введении его предшественника - Г-дофа, что и имеет место у больных дофа-зависимой дистонией. Подтверждением патогенетической значимости выявленных мутаций в гене ГЦГ-1 является значительное снижение уровня активности этого фермента (lt;20% от исходного уровня) у обследованных больных дофа-зависимой дистонией [Iehinose Н. et al., 1994; Hirano М. et al., 1998].
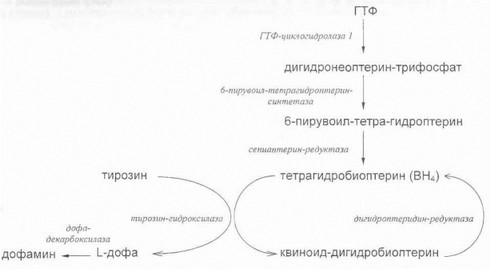
Рнс. 51. Биохимический путь синтеза дофамина
ДНК- диагностика наследственных болезней ... 229
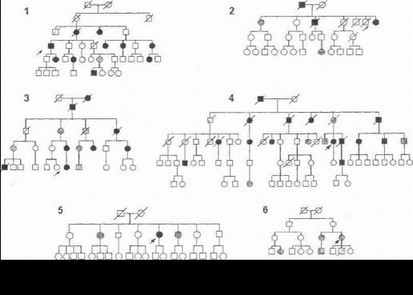
К настоящему времени идентифицировано свыше 40 различных (преимущественно толковых) мутаций в кодирующей области гена ГЦГ-1 у больных из различных популяций мира [Ichinose Н. et al., 1994; 1995; Bandmann О. et al., 1996; 1998; Furukawa Y. et al., 1996; Jarman P. et al., 1997; Hirano M. et al., 1998; Steinberger D. et al., 1998]. Нами впервые был проведен мутационный скрининг гена ГЦГ-1 в российских семьях с дофа-зави- симой дистонией [Маркова Е.Д. и др., 2000; Illarioshkm S. et al., 19981. На рис. 52 показаны наиболее информативные родословные семей с дофа-зависимой дистонией, обследованных в нейрогенетическом отделении НИИ неврологии РАМН. Нами выявлены 5 толковых мутаций в гене ГЦГ-1 (таблица 5), четыре из которых являются новыми и не описанными ранее в других популяциях.
Таблица 5
Идентифицированные мутации в гене ГЦГ-1 у больных дофа-зависимой дистонией в российских семьях
|
Э кзо и |
II у к л е оти д и а я замена |
Биологический эффект мутации |
|
1 |
ATG -gt; AAG |
М е11 02Lys |
|
1 |
АС G -Д AAG |
Thr94Lys |
|
2 |
TGT -Д TGG |
Cys141T rp |
|
4 |
AGT -Д ACT |
Seri 76Thr |
|
6 |
С G А-gt; TG А |
Arg21 6stop |
Таким образом, наши данные подтверждают мнение других авторов об уникальном характере мутаций в абсолютном большинстве случаев аутосомно-доминан- тной дофа-зависимой дистонии. В связи с этим прямая ДНК-диагностика данного заболевания требует проведения тотального скрининга всей кодирующей области гена. Наиболее общепринятым подходом является проведение SSCP-анализа отдельных экзонов гена с последующим прямым секвечированием образцов с аномально подвижными фрагментами ДНК. На рис. 53 показан пример такой Д11К-диагносгики.
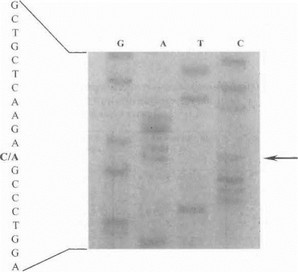
Рис. 53. Идентификация мутации 281 С^А в первом экзоне гена ГЦГ -1 (прямое секвенирование)
Стрелкой обозначена област ь муки щи (нуклеотидная замена С—gt;А j
Учитывая сравнительно небольшой размер кодирующей области гена (6 коротких экзонов), некоторые исследователи предпочитают проводить непосредственное секвенирование всей кодирующей области, не прибегая к SSCP-анализу.
Многие авторы отмечают, что выявляемые мутации в кодирующей области гена ГЦГ-1 обусловливает лишь половину всех случаев дофа-зависимой дистонии [Ichinose Н. et al., 1994; Bandmann О. et al., 1996; 1998; Furukawa Y. et al., 1996]. Данный факт принято объяснять генетической гетерогенностью данной формы дистонии либо возможной локализацией повреждений в некодирующих участках ГЦГ-1 (промоторная зона, инт- ронные мутации с нарушениями сплайсинга и др.) [Furukawa Y., Kish S., 1999]. По нашим данным, в российских семьях с несомненным аутосомно-доминантным наследованием болезни мутации в гене ГЦГ-1 обнаруживаются более чем в 80% случаев. Напротив, в спорадических случаях и при возможном аутосомно-рецессив- 1юм наследовании дофа-зависимой формы дистонии мутации ГЦГ-1 крайне редки. Это позволяет склониться в пользу точки зрения о роли других генов в развитии основной части спорадических и аутосомно-рецессив- ных случаев дофа-зависимой дистонии (см. далее).
5-летний опыт молекулярного анализа гена ГЦГ- I в семьях различного этнического происхождения показал, что у небольшой части гетерозиготных носителей мутации заболевание может проявляться не классическим генерализованным фенотипом ригидной формы ди- с, онии, а разнообразными атипичными клиническими нариантами - такими как фенотипы атетоидного церебрального паралича и спастической параплегии, паркинсонизм (в том числе паркинсонизм с Г-дофа-индуциро- иа иными дискинезиями), фокальная дистония конечностей, оромандибулярная дистония [Bandmann О. et al., 1996; 1998; Steinberger D. etal., 1998; Steinberger D. etal., 1999; Tassin J. et al., 2000]. Мы наблюдали больных, у которых единственным проявлением носительства мутантного гена ГЦГ-1 была непостоянная эквиноварусная поза стоп (главным образом, при ходьбе) либо изолированный статокинетический тремор рук [Markova Е. et al., 1999]. Во всех указанных случаях клиническая симптоматика значительно регрессировала после назначения малых доз L-дофа. Таким образом благодаря ДНК-тес- тированию стало понятным, что клинические проявления дофа-зависимой дистонии являются чрезвычайно вариабельными, а диагностические рамки данной формы наследственной дистонии - весьма широкими. Это- позволяет говорить о гораздо более высокой пенетрант- ности мутантного гена дофа-зависимой дистонии, чем традиционно цитируемая цифра 35-40% [Nygaard Т. et al., 1988]. Ранее при анализе пенетрантности явно недооценивалась роль различных атипичных и «стертых» случаев болезни, поскольку без ДНК-диагностики генетический статус таких лиц не мог быть определен с достоверностью. В последних работах авторы включали в сегрегационный анализ всех выявленных носителей мутаций в гене ГЦГ-1, имеющих даже минимальные клинические проявления, в результате значение пенетрантности гена оказалось равным 80-100% [Steinberger D., et al., 1998], что близко и к нашей оценке (gt;70%).
Интересно отметить, что наличие мутаций ГЦГ-1 в гомозиготном состоянии ведет к манифестации совершенно особого заболевания - атипичной неонатальной гиперфенилаланинемии [Blau N. et al., 1995; Furukawa Y., Kish S., 1999]. Оно обусловлено блоком активности тирозин-гидроксилазы (см. рис. 51), глубоким сочетанным дефицитом всех катехоламинов, а так-
же повышением содержания в крови фенилаланина (последнее связано с тем, что при выраженной недостаточности тетрагидробиоптерина страдает не только тиро- зин-гидроксилаза, но и фенилаланин-гидроксилаза). Клинически данный синдром характеризуется неонатальной задержкой двигательного развития, гипотонией, эпилептическими припадками, умственной отсталостью. У некоторых больных при особой комбинации мутантных аллелей гена ГЦГ-1 наблюдается «промежуточный» фенотип болезни с задержкой психомоторного развития и речи, прогрессирующей дистонией и умеренным положительным эффектом при назначении L-дофа [Furukawa Y. et ak, 1998]. Было показано, что при синдроме атипичной гиперфенилаланинемии имеет место практически нулевая активность ГЦГ-1, при «промежуточной» форме она обычно не превышает 1—2%, у больных дофа-зависимой дистонией она составляет 5-20%, а при значениях активности фермента выше 30- 40% какие-либо клинические проявления недостаточности ГЦГ-1 отсутствуют [Ozelius L., Breakefield X., 1994; Furukawa Y. et ak, 1998; Furukawa Y., Kish S., 1999]. Таким образом,, заболевания, обусловленные патологией гена ГЦГ-1, представляют собой особый континуум взаимно перекрывающихся состояний - от «стертых» субклинических форм дофа-зависимой дистонии па одном конце спек- [ ра до тяжелой неонатальной гиперфенилаланиемии на другом; при этом степень тяжести клинического синдрома увеличивается по мере нарастания дозы мутантного гена и дефицита активности фермента ГЦГ-1.
Сравнительно недавно были открыты два новых самостоятельных аутосомно-рецессивных варианта дофа- ’.ависимой дистонии, связанных с повреждением других I енов биохимической цепи синтеза дофамина (см. рис 51). Один из них обусловлен мутациями в гене 6-пирувоил-тетрагидроптерин-синтетазы (6-ПТГС), катализирующей вторую реакцию в указанной биохимической цепи [Hanihara Т. et al., 1997]. До настоящего времени описана лишь одна больная с гомозиготной мутацией в гене 6-ПТГС, имевшая «мягкую» генерализованную симптоматику дофа-зависимой дистонии с типичными дневными флюктуациями симптомов [Hanihara Т. et al., 1997]. Второй вариант аутосомно-рецессивной дофа-зависимой дистонии обусловлен мутациями в гене тирозин-гидроксилазы, локализованном на хромосоме llpll.5 [KnappskogP. etal., 1995; Ltidecke В. et al., 1995], что приводит к нарушению функции данного фермента и блоку синтеза дофамина. Назначение этим больным L-дофа также оказывает выраженный положительный эффект. По сравнению с аутосомно-доминантной дофа- зависимой дистонией, при дистонии с дефититом тирозин-гидроксилазы более часто наблюдается развитие синдрома паркинсонизма (в том числе в самом дебюте болезни) и несколько реже - дневные флюктуации тяжести симптомов [Furukawa Y., Kish S., 1999]. Очевидно, что исследование активности ферментов 6-ПТГС и ти- розин-i идроксилазы должно рассматриваться в качестве важных диагностических биохимических тестов в ауто- сомно-рецессивных и спорадических случаях дофа-зависимой дистонии.
Дофа-независимая торсионная дистония. Основной ген дофа-независимой дистонии, получивший обозначение DYT1, был картирован в 1989 году в хромосомном локусе 9q32-34 [Ozelius et al., 1989]. Локализация мутантного гена в области 9q34 была подтверждена в большинстве семей с аутосомно-доминантной дофа- независимой генерализованной дистонией в различных этнических группах мира [Kramer Р. et al., 1990; 1994]. Анализ серии DYT1-сцепленных высокополиморфных маркеров показал, что в популяции евреев ашкенази (характеризующейся очень высокой частотой данной формы дистонии) у больных отмечается один и тот же характерный гаплотип, свидетельствующий о едином генетическом происхождении дофа-независимой дистонии в данной этнической группе |Bressman S. et al., 1994; RischN. et al., 1995].
В 1997 ген DYT1 был идентифицирован [Ozelius L. et al.. 1997]. Его продукт - неизвестный ранее белок торсин А, имеющий определенную гомологию с семейством белков «теплового шока», играющих роль в предохранении клеток ох термического стресса. Эти белки имеют АТФ-связывающую активность и, помимо регуляции термотолерантпости, могут участвовать в процессах образования везикул, конформационных изменениях белков, регуляции клеточных сигналов и функционировании митохондрий [Parsell D., Lindquist S., 1993; Ozelius L. et al., 1997]. Augood S. с соавторами показали, что торсин А экспрессируется в дофаминергических нейронах компактной части черной субстанции, а также в мозжечке и гиппокампе [Augood S. et al., 1998]. В совокупности вышеуказанные данные свидетельствуют о возможной роли торсина А в процессах высвобождения нейромедиаторов в ынгро- стриарной системе мозга и/или поддержании энергетического потенциала дофаминергических нейронов и иейронопротекции.
11а сегодняшний день единственной мутацией, выявленной в гене DYT1 у больных дофа-независимой формой торсионной дистонии, является делеция трех нуклеотидов GAG в 946-м положении 5-го экзона гена (в гетерозиготном состоянии). Эта делеция ведет к утрате аминокислоты глутамата в карбоксильной части белка. Высокая частота данной мажорной мутации описана у больных самых различных национальностей в популяциях Европы и Северной Америки [Ozelius L. et al., 1997; Valente E. et al., 1998; Lebre A. et al., 1999; Kamm C. et al., 1999]. Если в этнической группе евреев ашкенази преобладающее носительство делеции GAG у больных обусловлено в основном «эффектом основателя» и наследованием общей предковой мутантной хромосомы, то в других этнических группах данная деления возникает повторно в результате многократных независимых мутационных событий. Последнее обстоятельство подтверждается наличием у этих больных разных гаплогипов по маркерам 9q34, сцепленным с мутантной хромосомой [Lebre A. et al., 1999; Kamm С. et al., 1999|. Возникновение делеции GAG de novo было подтверждено в 2 семьях, исследованных нами совместно с американскими исследователями из Гарвардского Университета [Klein С. et al., 1998 (а)]. Многократное возникновение одной и той же мутации у различных больных является сравнительно редким феноменом в клинической нейрогенетике. При дофа-независимой дистонии ЭТо связано с существованием в гене DYT1 тандемного 24-нуклео- тидного повтора в виде двух не полностью гомологичных копий, включающих GAG-димер; делеция первого GAG-элемента данного димера приводит к существенному повышению степени гомологии соседних повторов, «провоцируя» возникновение данной мутации [Klein С. et al., 1998 (a)].
Анализ клинико-генетических корреляций показал, что универсальная делеция GAG в гене DYT1 обнаруживается не только у больных с типичной генерализованной формой дофа-независимой дистонии с ранним дебютом. В некоторых семейных случаях данная мутация была выявлена также у больных с фокальными (писчий спазм), мультифокальными и сегментарными формами дофа-нечувствительной дистонии, а также у больных с атипичными клиническими проявлениями болезни (постуральный тремор рук, заикание вследствие дистонии оральной мускулатуры и т.п.) [Миклина Н.И., 1999; Gasser Т. et al., 1998 (б); Lebre A. et al., 1999; Kamm C. et al., 1999; Slommsky P. et al., 1999]. Делеция GAG при указанных вариантах заболевания обнаруживается весьма редко, однако данный факт позволяет существенно расширить спектр клинических проявлений 9р34-сцепленной формы наследственной дистонии и должен приниматься во внимание при медико-генетическом консультировании. В ряде случаев делеция GAG в изучаемой области гена DYT1 была выявлена также у больных, не имевших семейного анамнеза [Ozelius L. et al., 1997; Valente E. et al., 1998; Slominsky P. et al., 1999; I Iras sat D. et al., 2000]. Большинство указанных случаев, по-видимому, объясняются неполной пенетрантносхъю мутантного гена [Brassat D. et al., 2000] либо отсутствием достоверной генеалогической информации.
В нейрогенетическом отделении ЫИИ неврологии РАМН в 1998-2000 гг. обследовано 46 больных с различными клиническими вариантами дофа-независи- 'мой дистонии, включая как семейные, так и спорадические случаи заболевания [Миклина Н.И., 1999; Маркова Е.Д. и Др., 2000; Slominsky Р. et al., 1999]. Обобщенный анализ результатов тестирования больных на носительство делении GAG в гене DYT1 представлен На рис. 54. Суммарно данная мажорная делеция выявлена нами у 28 больных из 18 семей (69,2% обследованных российских семей с дофа-независимой дистонией). Как видно на рис. 54, большинство случаев с идентифицированной делецией составляет классическая генерализованная дофа-нечувствительная дистония с ранним началом симптомов (55%) и фокальная дистония конечностей (36%), а у небольшой части носителей мутации
заболевание манифестировало в виде атипичных «стертых» форм (9%). В группе больных с отрицательными результатами ДНК-диагностики генерализованная дистония наблюдалась лишь у 25% больных, тогда как буль- шую часть случаев составили фокальные и атипичные формы заболевания.

Рис. 54. Частота делеции GAG в гене DYT1 при различных клинических вариантах дофа-независимой дистонии
Таким образом, согласно нашим данным, основным показанием для проведения ДНК-анализа на носи- тельство делеции GAG в гене DYT1 является клиника генерализованной или фокальной дистонии, нечувствительной к препаратам L-дофа, особенно при манифестации симптомов на первом десятилетии жизни. По мнению ряда авторов, в спорадических случаях генерализованной дистонии анализ DYT1-делеции следует проводить при дебюте болезни до 26 лет, манифестации симптомов с мускулатуры конечностей и развитии генерализованной формы дистонии [Brassat D. et а].. 2000; Bressman S. et al., 2000].
Обнаружение универсальной делеции GAG в гене DYT1 у значительной части больных дофа-независимой дистонией дает возможность проведения нростой и достоверной ДНК-диагностики в большинстве семейных и части спорадических случаев заболевания, а также у клинически здоровых родственников из группы риска в консультируемых отягощенных семьях. На рис. 21 (с. 79) показана типичная электрофореграмма полиакриламидного геля, на которой у носителей делеции четко определяется более короткий фрагмент амплифицированной ДНК. Делеция GAG приводит к исчезновению сайта рестрикции для рестриктазы BseKl, поэтому во многих лабораториях для прямой ДНК-диагностики делеции используется обработка продуктов амплификации ферментом AstRI с последующим электрофорезом ДНК в агарозном геле (рис. 55). По нашему мнению, второй метод прямой ДНК-диагностики болезни является предпочтительным, поскольку он более специфичен и позволяет абсолютно точно диагностировать именно искомый три- пуклеотидный GAG-дефект в изучаемой области гена.
До настоящего времени не описан ни один больной с дофа-независимой дистонией, который бы имел другие мутации в гене DYT1 (помимо делеции GAG). 11е исключено, что другие мутации в гене DYT1 либо являются «клинически немыми», либо приводят iw совершенно иному фенотипу болезни, либо, наконец, яв- ияются эмбрионально летальными и несовместимы с жизнью [Ozelius L. et al., 1997]. Ответ на этот вопрос, который по-видимому будет получен в самое ближайшее время, чрезвычайно важен как для дальнейшего совершенствования генодиагностики дофа-независимой формы дистонии, так и для более глубокого понимания патофизиологии дистоничсского гиперкинеза.
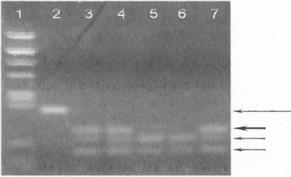
Рис. 55.Прямая ДНК-диагностика дофа-независимой дистонии с помощью рестрикционного анализа Продукты амплификации 5-го экзона обработаны рестриктазой Bse RI. Дорожка 1 -маркер, дорожка 2 -контроль (нативный продукт амплификации без рестрикции, указан длинной стрелкой), дорожки 3,4,7- больные дофа-независимой дистонией, имеющие делению GAG в гене DYT1, дорожки 5 и 6 - больные без делеции GAG. Короткой жирной стрелкой указан мутантный аллель (более длинный рестрикционный фрагмен т), короткими тонкими стрелками -продукты рестрикции нормального аллеля.
Известна особая «смешанная» форма аутосомно- доминантной дистонии, детальное описание которой поедставлено С Klein с соавторами [Klein С. etal., 1998 (б)]. В семьях отмечается неполная пейетрантность мутантного гена, раннее начало болезни и отсутствие чувствительности к препаратам L-дофа, что сближает данное заболевание с DYT1-ассоциированной дистонией. Однако в отличие от DYT1-дистонии при «смешанной» наследственной дистонии у большинства больных развиваются фокальные формы заболевания с преимущественным вовлечением краниоцервикальной мускулатуры (шея, лицо, артикуляционная мускулатура), тогда как генерализация процесса имеет место лишь у небольшого числа больных.
В 1997 г. в результате исследования 2 семей с данной формой дистонии ген заболевания (локус DYT6) был локализован на хромосоме 8p21-q22 [Almasy L. et al., 1997]. Таким образом была подтверждена генетическая гетерогенность дофа-печувствительной формы торсионной дистонии. В соответствующих информативных семьях при исключении у больных носительства деле- ции GAG в гене DYT1 возможен анализ сцепления с локусом DYT6 на 8-й хромосоме и (в случае подтверждения сцепления) проведение косвенной Д1IK-диагностики в группе риска.