
Понятием «первичный паркинсонизм» объединяются дегенеративные заболевания головного мозга, характеризующиеся прогрессирующей избирательной ш- белью нейронов нигро-стриарной системы и развитием изолированного синдрома паркинсонизма (тремор покоя, мышечная ригидность, одиго-брадикинезия). Согласно современным представлениям, выделяют две основные нозологические формы первичного паркинсонизма - болезнь Паркинсона и ювенильный паркинсонизм [Шток В.Н. и др., 1998]. В развитии первичного паркинсонизма значительную роль играют наследственные факторы, в связи с чем молекулярно-генетический анализ стал в последние годы одним из наиболее приоритетных направлений в исследовании данных заболеваний.
Болезнь Паркинсона (семейные формы). Болезнь Паркинсона представляет собой второе по частоте (после болезни Альцгеймера) нейродегенеративное заболевание человека, которое встречается во всех популяциях мира и имеет распространенность 2-4% среди лиц старше 65 лет [De Rijk М. et al., 1995]. Заболевание характеризуется типичным тремором покоя рук и других частей тела частотой 4-6 Гц, экстрапирамидной мышечной ригидностью, брадикинезией, постуральной неустойчивостью, вегетативными нарушениями, постепенно присоединяющимися когнитивными расстройствами подкоркового типа и неуклонно прогрессирующим течением [Голубев В.Л. и др., 1999; Marsden C.D., 1994]. Морфологическая картина болезни Паркинсона включает дегенерацию и гибель дофаминергических нейронов компактной части черной субстанции, а также наличие в сохранных клетках особых интранейрональных эозинофильных включений - телец Леви [Gibb W., Lees А., 1988]. Использование L-дофа (предшественник дофамина) и других препаратов, корригирующих характерные для болезни Паркинсона центральные нейротрансмит- терные нарушения, позволяет значительно улучшить двигательную активность и качество жизни больных на
протяжении ряда лет. Однако современная терапия не предотвращает дальнейшего неуклонного прогрессирования болезни и появпения большого числа тяжелых осложнений, многие из которых являются следствием проводимого лечения [Голубев В.Л. и др., 1999; Marsden C.D., 1994].
Большинство случаев болезни Паркинсона являются спорадическими. У этих больных заболевание имеет мультифакториальную природу с отчетливой генети ческой предрасположенностью, о чем подробно говорится в главе 4. В то же время в литературе неоднократно описаны семейные формы болезни Паркинсона, когда заболевание имело место у нескольких родственников из одного или разных поколений [Spacey S., Wood N., 1999]. По различным оценкам, частота таких семейных форм составляет в среднем около 10-20% всех случаев болезни Паркинсона [Marder К. et al., 1996; Duvoisin R., 1998; Gasser T., 1998; JElbaz A. cl al., 1999]. Особенно значимо роль наследственности выступает при анализе редко встречающихся больших семей, в которых болезнь Паркинсона наблюдается у большого числа родственников и наследуется как монотонный признак; в большинстве таких описанных родословных отмечен аутосомпо- доминантный тип наследования с неполной пенетрант- ностью мутантного гена [Duvoisin R., 1998; Spacey S., Wood N., 1999] Таким образом, в некоторых случаях развитие болезни Паркинсона определяется повреждением одного основного гена. Для этих случаев применимы все основные подходы к медико- генетическому консультированию и ДНК-диагностике, которые используются в семьях с другими наследственными моногениы- ми болезнями, рассматриваемыми в данной главе.
В 1997-1999 гг. были открыты 4 генетических локуса, ответственных за развитие аутосомио-доминантных случаев болазни Паркинсона, причем для двух из них идентифицированы мутантные гены. Первый из них расположен на хромосоме 4q21-23 (данный локус обозначен символом PARK1) и кодирует синтез а-синуклеина - основного белкового компонента телец Леви [Polymeropoulos М. et al., 1997]. К настоящему времени, несмотря на проведенное в мире обследование сотен семей с болезнью Паркинсона, описаны лишь 2 толковых миссенс-мутации в гене а-синуклеина: одна из них обнаружена в небольшом числе итальяно-греческих семей, имеющих общее происхождение из одного средиземноморского региона [Polymeropoulos М. et al., 1997; Athanassiadou A. et al., 1999], а вторая - в семье этнических немцев [Kruger R et al., 1998]. Обе мутации предположительно приводят к изменению пространственной организации а-синуклеина, следствием чего может являться нарушение его процессинга или деградации, приводящее к формированию амилоидоподобных белковых агрегатов [Spacey S., Wood N., 1999].
Нами было проведено прямое секвенирование гена а-синуклеина в 9 российских семьях с несомненным аутосомно-доминантным наследованием болезни Паркинсона (рис. 58), при этом мутаций в кодирующей области а-синуклеина выявлено не было. Результаты исследований различных авторов и наши собственные данные свидетельствуют о том, что мутации гена а-синуклеина представляют собой весьма редкую причину семейных форм болезни Паркинсона [Vaughan J. et al., 1998; Scott W. et al., 1999].
Следующий ген аутосомйо-доминантной болезни Паркинсона был выделен в 1998 году. Он локализован на хромосоме 4р14 (локус PARK5) и кодирует синтез убиквитин-карбокси-терминальной гидролазы L1 (UCH-L1) [Leroy Е. et al., 1998]. Данный белок составляет 1-2% от всего белкового состава мозга, обнаружи-
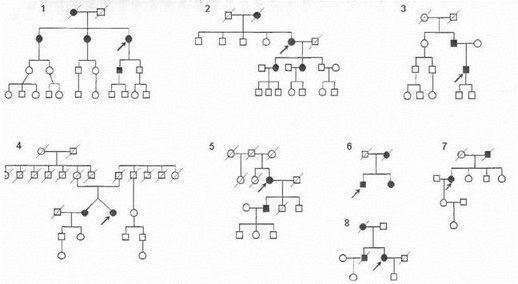
ис. 58. Примеры родословных обследованных семей с аутосомно-доминантной болезнью Паркинсона
ДНК- диагностика наследственных болезней ... 269
вается (как и а-синуклеин) в тельцах Леви и играет важную роль в процессах протеолиза полимерного убикви- тина. Предварительные данные свидетельствуют о том, что частота мутаций в гене иСП-Ll среди всех семейных случаев болезни Паркинсона чрезвычайно низка [Lincoln S. et al., 1999], однако в целом генетика данной молекулярной формы болезни Паркинсона остается малоизученной.
Еще один хромосомный локус (PARK3), ассоциированный с развитием аутосомно-доминантной болезни Паркинсона, картирован на хромосоме 2р13 [Gasser Т. et al., 1998а]. Пенетрантность мутантного гена в исследованной немецкой семье составила около 40%, что позволяет обсуждать роль локуса PARK3 в развитии определенной части спорадических случаев болезни Паркинсона. Наконец, еще в одной обширной родословной М. Farrer с соавторами в 1999 году обнаружили сцепление локуса болезни Паркинсона (PARK4 ) с хромосомной областью 4р 14—16.3; в данной семье (как отмечалось в разделе 3.2.4.1) «мутантный» гаплотип был ассоциирован не только с болезнью Паркинсона, но также со случаями «чистого» эссенциального тремора [Farrer М. et al., 1999]. Указанное наблюдение может свидетельствовать об определенной этиологической взаимосвязи эссенциального тремора и, как минимум, одной из генетических форм болезни Паркинсона. В литературе описаны также семьи с аутосомно-доминантной болезнью Паркинсона, в которых все вышеперечисленные генетические локусы были исключены [Gwinn- HardyK. et al., 2000].
Таким образом, уже сейчас вполне очевидно многообразие генетических вариантов семейного идиопатического паркинсонизма. По-видимому, развитие заболевания может быть обусловлено повреждением болыно- го числа генов и их белковых продуктов, взаимосвязанных между собой посредством участия в функционировании общих метаболических путей (например, регуляции процессов протеолиза, обеспечении нейронального белкового транспорта и т.п.). С этих позиций формирование амилоидоподобных белковых комплексов, входящих в состав телец Леви, может представлять собой универсальную патологическую реакцию как результат нарушенного на различных этапах процессинга нейрональных белков [Spacey S., WoodN., 1999].
Генетическая гетерогенность семейных случаев болезни Паркинсона существенно осложняет проведение ДНК-диагпостики в отягощенных семьях. Тотальный скрининг кодирующей области генов а-синуклеина и UCH-L1 является с практической точки зрения нецелесообразным ввиду чрезвычайной редкости мутаций в них при семейном паркинсонизме. То же самое, по-видимому, будет относиться и к каждому другому конкретному гену, когда они станут доступными для прямого анализа. Одним из путей повысить эффективность ДНК- тестировапия является выделение отдельных подгрупп больных, в каждой из которых особенности клиники могли бы помочь правильному выбору генов для мутационного скрининга. 11апример, для мутаций в локусе PARK.1 (ген а-синуклеипа) характерно более раннее начало и быстрое прогрессирование болезни [Polymeropoulos М. et al., 1997], для РА11К4-ассоцииро- вапной формы (как уже было отмечено) характерно сочетание в семье случаев болезни Паркинсона и эссенци- ального тремора [Farrer М. et al., 1999] и т.д. Сейчас мы находимся лишь на самом начальном этапе анализа клинико-генетических корреляций и определения характерного фенотипа отдельных генетических форм болезни Паркинсона. Дальнейшее накопление и обобщение этих данных на основе широкого международного сотрудничества позволит выработать более четкий алгоритм для проведения ДНК-диагностцки и медико-генетического консультирования в семьях, отягощенных наследственными случаями болезни Паркинсона.
Ювенильный паркинсонизм. Ювенильный паркинсонизм представляет собой особую наследственную форму первичного паркинсонизма, детально изученную лишь в последние 10-15 лет [Narabayashi Н. et al., 1986; Takahashi Н. et al., 1994; Ishikawa, Tsuji S., 1996; Tassin J. etal., 1998]. Заболевание распространено повсеместно и несколько чаще встречается у женщин. Тип наследования в большинстве описаний - аутосомно-рецессивный, в связи с чем многие случаи ювенильного паркинсонизма в небольших по размеру семьях являются спорадическими. Дебют симптомов чаще всего приходится на 2—4-е десятилетие жизни, реже на более ранний возраст. Клиническая картина ювенильного паркинсонизма имеет ряд особенностей по сравнению с классической болезнью Паркинсона. Главными из них являются: частое сочетание паркинсонизма с дистонией; наличие флюктуаций в выраженности симптомов паркинсонизма и дистонии на протяжении дня ('ухудшение состояния к вечеру, улучшение утром или после дневного сна); отсутствие (даже при многолетнем течении болезни) деменции и других психических расстройств и, наоборот, частое сочетание паркинсонизма с повышением сухожильных рефлексов и другими пирамидными симптомами; прекрасный многолетний терапевтический эффект небольших доз L-дофа (в среднем 200-250 мг L-дофа в сутки в комбинации с карбидофой), который, однако, нередко осложняется присоединением размашистых дискинезий и усилением выраженности симптомов дистонии. Течение заболевания медленно прогрессирующее (на протяжении десятилетий), прогноз относительно благоприятный.
Патологанатомическая картина ювенильного паркинсонизма характеризуется отсутствием телец Леви в нейронах структур среднего мозга (это главное отличие от классической болезни Паркинсона) и гибелью нейронов с реактивным глиозом в компактной части черной субстанции и в области locus ceruleus [Takahashi Н. et al., 1994].
Основной ген аутосомно-рецессивного ювенильного паркинсонизма картирован в хромосомной области 6q25.2-27 (локус PARK2) [Matsumine Н. et al., 1997]. Данный ген был выделен в 1998 году: он содержит 12 экзонов и кодирует новый белок паркин, состоящий из 465 аминокислот [Kilada Т. et al., 1998]. В нейроне паркин локализован в комплексе Гольджи и цитозоле; им- муногистохимическим методом показано, что у больных ювенильным паркинсонизмом паркин отсутствует во всех отделах головного мозга [Shimura et al., 1999]. Точная функция паркина в клетке до настоящего времени не установлена. У больных ювенильным паркинсонизмом выявлено большое число различных мутаций в гене паркина, представленных как ггруктурными перестройками (дслеции, реже *1тцликании различной протяженности), так и точковыми мутациями [Hattori N. et al., 1998; Lucking C. et al., 1998; 2000; Abbas N. et al., 1999].
Специфический характер мутаций в гене паркина определяет некоторые особенности методов мутационного скрининга у больных ювенильным паркинсонизмом. На первом этапе (особенно в инбредньтх семьях, в которых больные с высокой вероятностью являются носителями 2 копий идентичной мутантной хромосомы) целесообразно исключить гомозиготную делецию того или иного участка гена. Для этой цели проводится амплификация отдельных экзонов гена (в том числе в мультиплексной ПЦР), после чего на электрофореграмме гомозиготная деления диагностируется на основании отсутствия ПЦР-продукта соответствующего экзона или группы экзонов. На следующем этапе анализа может быть использован специально разработанный протокол полуколичественной мультиплексной амплификации, позволяющий исследовать дозу гена и диагностировать гетерозиготные делеции или дупликации исследуемых экзонов [Lucking С. et al., 2000]. Наконец, для диагностики толковых мутаций в оставшихся случаях проводится секвенирование кодирующей области гена (в том числе после предварительного SSCP-анализа) на образцах геномной ДНК или кДНК. Комбинированное использование указанных методов ДНК-диагностики позволяет выявлять мутации в гене паркина более чем в 70% семейных и спорадических случаев ювенильного паркинсонизма, причем чаще всего мутации выявляются в группе больных с началом болезни до 20 лет [Lucking С. et ak, 1998; 2000; Abbas N. et al., 1999].
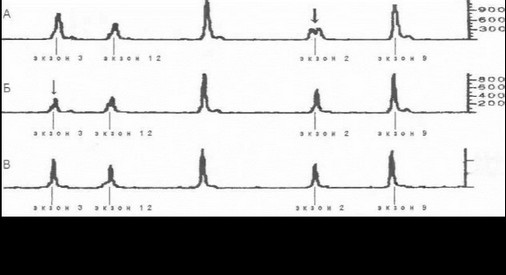
Под нашим наблюдением на протяжении 25 лет находится 6 семей с типичным аутосомно-рецессивным ювенильным паркинсонизмом, а также ряд изолированных случаев данного заболевания. Родословные двух из этих семей (в каждой из которых больны 3 сибсов) показаны на рис. 59. В 1999 году нами совместно с коллегами из Национального Института Здоровья и Медицинских Исследований Франции (INSERM, Париж) проведена первая у российских больных прямая ДНК-диагно- стика ювенильного паркинсонизма. В представленных на схеме родословных больные оказались компаунд-ге- терозиготами по различным делециям во 2-м, 3-м и 7-м экзонах гена паркина (рис. 60). Приводим краткие выписки из историй болезни пациентов в одной, из наблюдавшихся нами семей (родословная А на рис. 59).
Больная Тр. П-З, в возрасте 17 лет впервые отметила скованность в правой ноге и подволакивание нот
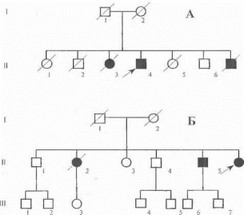
Рис. 59. Родословные обследованных семей с ювенильным паркинсонизмом
при ходьбе. Спустя несколько лет скованность распространилась па левые конечности, присоединилось дрожание рук. В 23 года вперьые обследована в НИИ неврологии РАМН, где после назначения небольших доз L-дофа (до 0,5 г в сутки в составе мадопара-125) был достигнут значительный терапевтический эффект: полностью исчезло дрожание, существенно уменьшилась выраженность экстрапирамидной ршидности, улучшилась походка. Спустя 6 лет на фоне непрерывного лечения появились отчетливые дискинезии пика дозы, выраженность которых постепенно нарастала, что стало существенно ограничивать самообслуживание. Последнее стационарное лечение в НИИ неврологии РАМН проводилось в возрасте 44 лет. В неврологическом ста
тусе на фоне отмены L-дофа отмечалась выраженная олигобрадикинезия, повышение мышечного тонуса по экстрапирамидному типу в конечностях (больше справа), среднеамплитудный ритмичный тремор покоя рук. Походка скованная, «шаркающая», в резко замедленном темпе, при ходьбе содружественные движения рук отсутствуют. Сухожильные рефлексы оживлены, без четкой разницы сторон. Гиперсаливация; речь монотонная, слабо модулированная. При назначении 1 таблетки на- кома наблюдалось исчезновение тремора рук и значительный регресс акинетико-ригидного синдрома, при этом на фоне максимального действия препарата в течение 30-40 минут появлялись размашистые хореоформ- ные гиперкинезы конечностей (особенно ног), подергивания головы и дискинезии мимических мышц. Наряду с этим периодически, вне четкой связи с временем приема L-дофа, отмечаются кратковременные эпизоды резко выраженных генерализованных гиперкинезов в виде баллистических движений конечностей, «подбрасывания» туловища на кровати; в момент гиперкинезов самостоятельная ходьба невозможна. При КТ патологических изменений в веществе головного мозга не выявлено. После «лекарственных каникул» больной назначена L-дофа (наком) в суточной дозе 0,75 г в 6 приемов в комбинации с паркопаном и клоназепамом; на фоне данной схемы лечения удалось практически полностью купировать дискинезии, больная продолжала прием препаратов на протяжении 12 лет со стабильным терапевтическим эффектом. В возрасте 56 лет скончалась от острого нарушения мозгового кровообращения.
Больной Тр. П-4, пробанд, в возрасте 23 лет стал отмечать скованность в ногах во второй половине дня и после умеренной физической нагрузки; скованность полностью проходила после сна. На протяжении нескольких лет скованность в ногах постепенно нарастала и распространилась на руки, больной стал отмечать дрожание рук, значительно изменилась походка, появились пропульсии. В возрасте 28 лет были впервые назначены препараты L-дофа (2 капсулы в день мадопара- 250) в сочетании с циклодолом (4 мг в день); спустя несколько дней от начала лечения скованность практически исчезла (она отмечалась в ногах лишь в утренние часы до приема Ь-дофа)Ь нормализовалась походка. С 29 лет наблюдается в ИИИ неврологии, на протяжении многих лет принимал препараты L-дофа в различных комбинациях с центральными холиноли гиками, без существенных побочных эффектов. Последнее стационарное обследование в Институте проводилось в возрасте 54 лет. Fla фоне отмены препаратов отмечается выраженное генерализованное повышение мышечного тонуса по э'кетрапирамидному типу, отчетливая брадикинезия, гипомимйя, тремор покоя рук средней интенсивности, уменьшающийся при целенаправленных движениях. Походка «шаркающая», при ходьбе отсутствуют синкинезии рук, отмечаются редкие пропульсии, постурааеная неустойчивость. Сухожильные рефлексы оживлены, симметричные; выявляются рефлексы орального автоматизма. На МР- томограммах головного мозга отмечается слабое расширение III и боковых желудочков. 11осле назначения адекватной схемы приема препаратов (1-дофа 0,375-0,5 I' в день в 2 приема в составе накома, циклодол 2 мг в день в 2 приема) отмечено значительное уменьшение выраженности симптомов паркинсонизма. Эффект сохраняется на протяжении многих Лет, больной проживает отдельно от семьи и полностью обслуживает себя.
Больной Тр. II-7, заболел в возрасте 19 лет, когда ¦ гала беспокоить скованность и утомляемость ног к концу дня. Через несколько месяцев присоединилась
скованность в мышцах рук, появилось дрожание в конечностях (больше слева), изменилась походка. С 21 года наблюдается в НИИ неврологии РАМН; в Институте впервые были назначены препараты L-дофа, которые больной с отличным эффектом принимал на протяжении многих лет (первоначально - чистую L-дофу в дозе до 3 г/сут, а в дальнейшем - в составе накома в дозе 0,5 г в день). Последний раз поступил для коррекции схемы лечения в возрасте 34 лет в связи с усилением тремора и ухудшением самообслуживания. В неврологическом статусе на фоне отмены препаратов отмечалась легкая гипомимия, значительное повышение мышечного тонуса по экстрапирамидному типу в конечностях (больше слева), выраженная генерализованная брадики- незия, мелкоамплитудный тремор покоя рук (Sgt;D). При ходьбе туловище напряжено, наклонено вперед, походка «шаркающая», мелкими шажками, ахейрокинез слева. Сухожильные рефлексы на руках оживлены, на ногах высокие, отмечается клонусоид стоп, рефлекс Россолимо с двух сторон, рефлексы орального автоматизма. Отмечаются изменения в эмоциональной сфере (эйфоричность, некоторая расторможенность), а также негрубое интеллектуальное снижение. При КТ головного мозга выявляется умеренное расширение ликворосодержащих пространств. Больному был назначен более дробный прием прежней суточной дозы накома в комбинации с цикло- долом до 6 мг в день, в результате чего полностью исчезло дрожание, значительно уменьшилась скованность, улучшилась ходьба и самообслуживание. В возрасте 42 лет больной скончался от прободной язвы желудка и желудочного кровотечения.
Молекулярно-генетическое исследование было проведено в 1999 году у пробанда: при анализе гена пар- кина в экзоне 2 выявлена делеция двух нуклеотидов AG в положении 202-203 (см. рис. 60 А). Диагноз: аутосом- но-рецессивный ювенильный паркинсонизм.
Таким образом, в представленных случаях у 3 больных в семье наблюдались практически все «классические» клинические признаки ювенильного паркинсонизма - раннее начало болезни, хороший терапевтический эффект сравнительно небольших доз L-дофа, сохранявшийся на протяжении многих лет, отсутствие деменции, наличие пирамидной симптоматики, наличие ранних L-дофа-индуцированных дискинезий (у больной II -3), а также суточных флюктуаций выраженности симптомов паркинсонизма (у больного П-4).
После того как появилась возможность достоверной молекулярной диагностики ювенильного паркинсонизма, было показано существование ряда атипичных случаев данного заболевания. Так, у отдельных больных - носителей мутаций в гене паркина заболевание в развернутой стадии было практически неотличимо от классической болезни Паркинсона, а первые симптомы могли появляться только после 50-летнего возраста [Tassin J. et al., 1998; Lucking C. et al., 2000]. В других наблюдениях у больных, имевших мутации в гене паркина, клиническая картина соответствовала дофа-зависимой форме торсионной дистонии [Abbas N. et al., 1999; Tassin J. et al., 2000]. Нередко у остальных больных в описанных семьях наблюдалась типичная клиника ювенильного паркинсонизма с ранним началом симптомов (что обычно является решающим фактором в клинической диагностике ювенильного паркинсонизма в обследуемой семье). С практической точки зрения, отмеченные наблюдения демонстрируют важность тщательного анализа клинической картины у всех больных родственников, а также необходимость проведения ДНК-анализа с целью правильной диагностики ювенильного паркинсонизма и его дифференцирования от ранних случаев болезни Паркинсона и других сходных заболеваний.
Второй локус аутосомно-рецессивного паркинсонизма (PARK6), отличпющегося от «классических» случаев ювенильного паркисонизма несколько более поздним началом (от 32 до 48 лет), отсутствием дистонии в начале болезни и отсутствием дневных флюктуаций, был совсем недавно картирован па хромосоме 1р35—36 [Valente Е. et al., 2001]. Заболевание характеризуется весьма медленным течением и стабильным многолетним эффектом леводопа-препаратов, а также частыми лево- допа-индуцированными дискинезиями. Соответствующий мутантный ген не установлен.На той же хромосоме картирован локус еще одной молекулярной формы аутосомно-рецессивного паркинсонизма (PARK7) |van Duijfl С. et al., 2001). В семьях с аутосомно-рецессив- ным паркинсонизмом, в которых не было выявлено мутаций в пене ггаркина, целесообразно исследовать сцепление с данными новыми локусами на хромосоме I р и в случае подтверждения сцепления - проводить косвенную ДНК-диагностику у братьев-сестер пробанда.
В единичных семьях с типичной клинической картиной ювенильного паркинсонизма описан аутосом- но-доминантный тип передачи болезни | Dwork A. et al., 1993; lshikawa A., IVtivatalceT., 1995]. Это свидетельствует о вовлечении в патогенез ювенильного паркинсонизма и дру1 их генов, белковые продукты которых могут взаимодействовать с паркином в сложных биохимических реакциях, имеющих отношение к обмену дофамина.