
Хорея (болезнь) Гентингтона - тяжелое дегенеративное заболевание головного мозга, распространенность которого в большинстве попgt;ляций мира составляет 4-10 случаев на 100 000 населения. Хорея Гентингтона наследуется по аутосомно-доминантному типу. Морфологической основой болезни является атрофия подкорковых узлов (главным образом полосатого тела и бледного шара), а также гибель нейронов и атрофия коры больших полушарий с развитием наружной и внутренней гидроцефалии и прогрессирующим снижением общей массы мозга [Hayden М., 1981].
Принято выделять две основных клинических формы заболевания - гиперкинетическую и акинетико- ригидную [Иванова-Смоленская И.А. и др., 1998 (а); Penney Г, Young А., 1998]. Гиперкинетическая форма является наиболее типичной и характеризуется началом болезни на 4-6 десятилетии жизни; у больных нарастают размашистые генерализованные хореические гипер- кинезы и деменция подкоркового типа, развивающиеся на фоне выраженных эмоционально-волевых нарушений. Постепенное развитие экстрапирамидной ригидности.
кахексии и вегатативно-трофических нарушений знаменует собой переход болезни в так называемую позднюю акинетико-ригидную форму, представляющую собой финал заболевания. Самостоятельное значение имеет первичный ювенильный акинетико-ригидпый вариант болезни Гентингтона (вариант Вестфаля), наблюдаемый в 5-10% случаев. У таких больных заболевание манифестирует на 1-2-м десятилетии жизни и характеризуется особенно неблагоприятным течением, с развитием мышечной ригидности, контрактур, изменений поведения и психики, эпилептических припадков, миоклоний, пирамидных симптомов, атаксии. Хореические гиперки- незы могут быть выражены незначительно или вообще отсутствовать, в связи с чем диагностика данною варианта болезни весьма затруднена. Ювенильный вариант Вестфаля наблюдается обычно при передаче мутантного гена от больного отца, особенно если наследование болезни по мужской линии происходило в нескольких поколениях подряд (так называемый эффект отцовской передачи») [IJaydcn М., 1981; Folste'n S., 1989]. Еще одним характерным для хореи Гентингтона феноменом, связанным преимущественно с мужской передачей мутантного Гена, является антиципация нарастание тя жести болезни и появление ее в более молодом возрасте в «нижних» поколениях родословной [Folstein S., 1989; Penney J., Young A., 1998].
Хорея Гентингтона характеризуется неуклонно прогредиентным течением. Больные обычно погибаю т через 15-20 лет от момента появления первых симптомов вследствие иптеркуррентных заболеваний, травм или суицидальных действий. При ювенильной акинетико- ригидной форме длительность болезни значительно меньше. Напротив, в случае манифестации симптомов после 60-70 лет заболевание прогрессирует медленно, а выраженная деменция развивается редко; такой клинический вариант с поздним началом и относительно благоприятным течением чаще наблюдается при наследовании мутантного гена от матери [Penney J., Young А.,
- .
Принимая во внимание выраженный фенотипический полиморфизм хореи Гентинггоиа, а также неспецифический характер изменений, выявляемых при использовании рутинных лабораторно-инструментальных методов исследования (расширение боковых желудочков и субарахноидального пространства больших полушарий при КТ и МРТ, депрессия a-ритма на электроэнцефалограмме), важной задачей является внедрение в практику методов ДНК-диагпостики данного заболевания.
В 1983 году ген хореи Гентипгтона был картирован на коротком плече 4-й хромосомы (локус 4р 16.3) в одном из самых первых успешных исследований генетического сцепления у человека [Gusella J. et al., 1983]. Это послужило основой для разработки косвенной пре- симптоматической и пренатальной ДНК-диагностики хореи Гентингтоыа в иыфопмагивных семьях [Евграфов О.В. и др., 1991; Иванова-Смоленская И.А. и др., 1992; Meissen G. et al., 1988]. Благодаря накопленному в мире многолетнему опыту такой диагностики и некоторым клинико-генетическим особенностям хореи Гентингто- на (позднее начало, практически полная пенстрантность мутантного гена, фатальное течение и др.) данное заболевание стало классической «моделью» для изучения различных аспектов медико генетического консультирования, основанного на ДНК-тестировании [World Federation of Neurology Research Committee and Research Group on Huntington’s chorea, 1 989; Huggins M. et al., 1990].
Спустя 10 лет после картирования локуса болезни Гентингтона данный ген был идентифицирован
Международной исследовательской группой под руководством J. Gusella [The 1 hmiington’s Disease Collaborative Research Group, 1993]. Ген имеет размер 180 кб, состоит из 67 экзонов и кодирует белок с молекулярной массой 348 кДа и неизвестной функцией, получивший название «гентингтин». В своем первом экзоне ген содержит тандемную последовательность (CAGjn-повторов, число которых в норме составляет обычно от 6 до 32, а на мутантных хромосомах - от 36 до 180 [Krcrner В. et ah, 1994]. Таким образом, сущность мутации при хорее Ген- тиштона заключается в экспансии внутривенных тандемных тринуклеотидных повторов. Экспансия повторов CAG имеет место в транслируемой области гена и приводит к синтезу аномального белка, содержащего пропорционально удлиненный полиглутаминовый участок (каждый триплет CAG соответствует одной аминокис лоте глутамина в полипептидной цепи). Предполагается, что в результате происходит формирование патологических полиглутамин-опосредованных связей мутантного гентингтипа с молекулами ряда беи ков ЦПС, что обусловливает его цитотоксичность и в конечном счете селективную гибель нейронов, характерную для хореи Гентингтона [Albin R., Tagle D., 1995; Ross С., 1995]. Такой патогенез болезни подтверждается выявлением в мозге больных хореей Гентингтона характерных внутриядерных включений, содержащих мутантный гентингтин и представляющих собой высокомолекулярные ами лоидоподобные агрегаты [Li Н. et ah, 1995]. Интересно отметить, что лица, имеющие «двойную дозу» мутантного гена (гомозиготные носители патологически удлиненного CAG-участка), клинически неотличимы от боль- пых-гетерозигот по экспансии повторов [Durr A. et ah,
- .
Анализ гена гентингтипа у представителей основных этнических групп мира показал, что 0,75% всех хромосом характеризуются числом CAG-повторов, «промежуточным» между нормой и заведомо патологическими значениями [Кгешег В. et al., 1994]. Такие аллели, по- видимому, представляют собой премутацию и служат (в случае дальнейшего нарастания числа повторов) постоянным источником полных мутаций de novo в популяции [Myers R. et al., 1993; Nance M., 1996]. Важно подчеркнуть, что аллели с числом CAG-повторов 36-39 являются «зоной неполной пенетрантности»: часть лиц с указанным числом повторов могут иметь типичные проявления хореи Генгтингтона, тогда как у других носителей таких аллелей заболевание не проявляется даже в самом преклонном возрасте - вплоть до 95 лет [Rubinsztein D. et al., 1996; Brinkman R. et al., 1997; McNeil S. et al., 1997]. Очевидно, что возможность развития заболевания при наличии хромосомы с числом CAG-повтооов 36-39 определяется взаимодействием разнообразных экзо- и эндогенных факторов, которые могут иметь как протективное, так и патологическое значение [Nance М., 1996]. В случае наследования хромосом с числом CAG-повторов gt;40 пенетрантность гена становится полной, и болезнь неизбежно проявится при условии достижения соответствующего возраста.
Прямая ДНК-диагностика хореи Гентингтона ос нована на амплификации с помощью ПЦР участка первого экзона гена, содержащего тринуклеотидный CAG- сегмент, с последующим электрофоретическим разделением продуктов амплификации. Наличие мутации диагностируется на основании выявления аномального удлиненного фрагмента ДНК, содержащего увеличенное число CAG-повторов. На рис. 46 показана типичная элек- трофореграмма агарозного геля, позволяющая полуко личественным экспресс-методом достоверно визуализй ровать заведомо мутантные аллели.
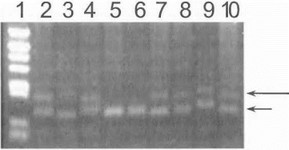
Рис. 46. Прямая ДНК-диагностика хореи Гентингтопа (электрофорез в агарозном геле)
Дорожка 1 -маркер, дорожки 2 4.7-10- больные хореей Гентннг- тона. дорожки 5,6 -контроль. Длинной стрелкой указан мутантный аллель (экспансия CAG-повторов гена гентинггина), короткой стрелкой - нормальный аллель.
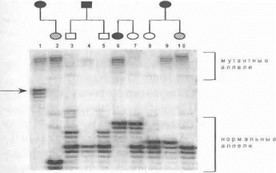
1'ис. 47. Прямая ДНК-диагностика хореи Гентингтопа ( шектрофорез в полиакриламидном геле)
Каждому члену семей, указанных над электрофореграммой, соответствует одна дорожка электрофореза. Дорожки 1.4, 6,9 - . юльные хореей Гентингтопа, дорожки 2и 10- носители мутации па пресимптоматической стадии (обозначены штриховыми i пмволами), дорожки 3, 5 7 8 - здоровые лица, нс являющиеся | к км [телями мутации. Стрелкой указан аллель с «промежуточным» • 11 ici I ом тринуклеотидных CAG-i ювторов.
При необходимости определения точного числа CAG-повторов (например, в случаях носительства «промежуточных» аллелей) ДНК-диагностика проводится с использованием полиакриламидного геля, обладающего значительно более высокой разрешающей способностью (рис. 47). Нейрогенетическим отделением НИИ неврологии РАМН в 1993-1999 г.г. накоплен уникальный опыт прямой ДНК-диагностики хореи Гентингтона в российской популяции. Нами обследовано gt;200 больных и их клинически здоровых родственников из группы риска (свыше 90 неродственных семей), что позволило сформулировать основные принципы медико-генетического консультирования и провести анализ разнообразных клинико-генетических корреляций при данном заболевании [Иллариошкин С.Н. и др., 1996 (а); Илла- риошкин С.Н., 1997; Иванова-Смоленская И.А. и др., 1998 (б); Клюшников С.А., 1998; Юношников С.А. и др.,
- . Было показано решающее значение прямого ДНК- анализа для подтверждения клинического диагноза хореи Гентингтона, а также для диагностики спорадических и атипичных вариантов заболевания (включая ранние и поздние акинетико-ригидные формы).
Обследование клинически здоровых родственников больных хореей Гентингтона позволило нам выделить группу из 25 носителей заведомо мутантного аллеля в возрасте от 18 до 48 лет (пресимптоматическая ДНК- диагностика). При использовании набора нейропсихо- логических тестов было установлено, что такие доклинические носители мутантного гена характеризуются рядом достоверных когнитивных и личностных изменений - повышением уровня реактивной и личностной тревожности, снижением объема памяти, нарушением практического и абстрактного мышления, нарушением концентрации внимания [Клюшников С.А., 1998]. Это позволяет сделать вывод о тонких изменениях в психо
логической сфере у носителей мутации задолго до манифестации развернутой картины хореи Гентингтона. Подтверждением ранних признаков когнитивной дисфункции у доклинических носителей мутантного гена хореи Гентингтона могут служить изменения, выявляемые у них при исследовании когнитивных вызванных потенциалов - в частности, достоверное удлинение латентности и снижение амплитуды волны РЗОО (рис. 48). Полученные нами данные существенно дополняют результаты ряда зарубежных авторов [Hahn-Barma V. et al., 1998; Kirkwood S. et al., 1999] и свидетельствуют о существовании при хорее Гентингтона достаточно продолжительной стадии «предболезни». Этот вывод имеет принципиальное теоретическое и практическое значение и должен учитываться при решении морально-этических проблем консультирования таких «асимптомных» лиц, а также (в будущем) при решении вопроса о назначении превентивной терапии.
У 42 обследованных нами клинически здоровых лиц из группы риска (дети и братья-сестры больных хореей Гентингтона) результат прямой ДНК-диагностики оказался отрицательным (число CAG-повторов в обоих аллелях гена lt;35). Таким образом, носительство мутации было отвергнуто, и данные лица были сняты с динамического наблюдения. В целом в российской популяции при исследовании свыше 600 хромосом нами ни в одном случае не было выявлено «пограничных» аллелей из «золы неполной пенетрантпости» (36-39 CAG- повторов), затрудняющих интерпретацию результатов ДНК-анализа. Это совпадает с данными других авторов об исключительной редкости таких аллелей гена ген- гингтина [Kremer В. etal., 1994; Rubinsztein D. etal., 1996]. И случае выявления носительства подобного «пограничного» аллеля сделать однозначное заключение о прогнозе ' консультируемого лица не представляется возможным.
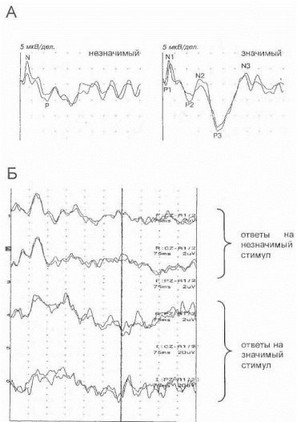
Рис. 48. Когнитивные вызванные потенциалы на пресимп- томатической стадии хореи Гентингтона
А. Норма (возраст испытуемого 25 лет). Показаны формы кривой в ответ на предъявление незначимого (слева) и значимого (справа) стимулов [Гнез- дицкийВ.В., 1997]. Б. Когнитивные вызванные потенциалы у 25-летнегlt;gt; клинически здорового носителя мутантного гена хореи Гентингтона (мутантный аллель содержит 44 копии САG-повторов). Отмечается удлинс ние латентности пика Р300 до 450 мс и снижение его амплитуды до 4.') мкВ (возрастные нормы - соответственно 320 мс и 9,4 мкВ).
Такие лица (как и их дети) должны оставаться в группе «высокого риска» и находиться под постоянным наблюдением врача-нейрогенетика.
Исследование гена гентингтина у больных с различными клиническими формами хореи Гентингтона дало нам возможность детально проанализировать взаимоотношения между генотипом и фенотипическими проявлениями болезни. Была установлена достоверная обратная корреляция между степенью экспансии CAG- повторов и возрастом, в котором манифестирую! первые сймптомы хореи Гентингтона (коэффициент корреляции г=-0,79, рlt;0,001). На рис. 49 показан график экспоненциальной зависимости между числом CAG-повто- ров и возрастом начала хореи Гентингтона, по которому можно ориентировочно рассчитать вероятный возраст дебюта заболевания у асимптомных носителей конкретного мутантного аллеля. Данная взаимосвязь была наиболее четко выражена для длинных аллелей (gt;52 повторов), тогда как при небольшой степени экспансии CAG- повторов возраст начала болезни варьировал в достаточно широких пределах, что следует учитывать при определении прогноза у консультируемых лиц из группы риска. В соответствии с выявленной закономерностью, все наблюдавшиеся нами случаи ювенильного акинетико- I нпидного варианта заболевания были связаны с наибольшей степенью тяжести генетического дефекта - число ('AG-повторов в мутантном аллеле у этих больных составило от 59 до 78. Обратная корреляция между степенью тяжести мутации и возрастом манифестации хореи I ептингтона, а также взаимосвязь акинетико-ригидного варианта Вестфаля с наиболее тяжелыми мутациями показаны также рядом зарубежных авторов на примере других популяций [Andrew S. etal., 1993; Duyao М. etal., 1993; Telenius H. et al., 1993; Trottier Y. et al., 1994].
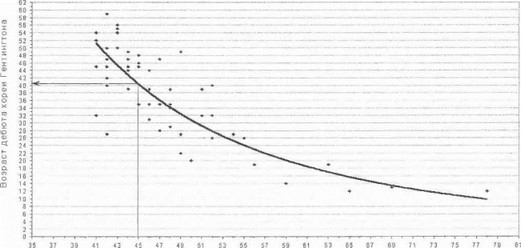
Число CAG-повторов
Рис. 49. Корреляция между числом тринуклеотидных CAG-повторов мутантного гена и возрастом манифестации симптомов хореи Гентингтона
На диаграмме показан пример прогностического расчета ориентировочного возраста, в котором может ожидаться начало заболевания у носителя мутантного гена с числом копий CAG-повторов 45.
Минимально выраженная экспансия CAG-повто- ров нередко обусловливает поздний и относительно доброкачественный фенотип заболевания с отсутствием выраженной деменции [Kremer В. et al., 1993; Britton J. et al., 1995]. Нами впервые была показана достоверная прямая корреляция между числом CAG-повторов в мутантном аллеле и темпом прогрессирования неврологических и психических симптомов хореи Гентингтона [Illarioshkin S. et al., 1994]. Как и при анализе возраста начала болезни, данная корреляция оказалась более строгой при длипе повтора, превышающей определенное «пороговое» значение (gt;50 С AG-триплетов). Таким образом, у носителей длинных, более «злокачественных» аллелей число копий CAG-повторов является ведущим фактором, определяющим дебют и прогпоз заболевания В то же время, при небольшой степепи экспансии CAG- повторов течение болезни является более вариабельным, что свидетельствует о влиянии на него у таких больных различных дополнительных молекулярных и/или экзогенных факторов.
Весьма важным является вопрос о характере нестабильности мутантного аллеля в зависимости от пола больного родителя, передающего патологический ген. 11аши данные по этому вопросу (рис. 50) полностью совпадают с результатами других авторов и свидетельствуют о том, что в большинстве случаев передачи мутации от больного отца и в трети всех случаев передачи мута- м,ни от больной матери у потомков наблюдается нарас- I ание числа копий CAG в мутантном гене [Duyao М. et al., 1993; Telenius Н. et al., 1993; Kremer В. et al., 1995]. 11лсяедование мутации от матери нередко сопровождается уменынепием числа повторов или отсутствием изменений в мутантном аллеле [Kremer В. et al., 1995].
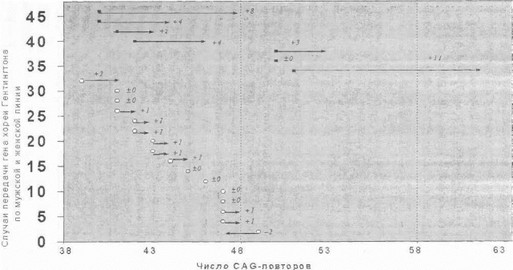
Рис. 50. Нестабильность мутантного аллеля при передаче генахореи Гентингтона по отцовской и материнской линии Черные квадратики — отцовская передача гена, белые кружки — материнская передача гена.
222 Глава 3
Крайняя степень экспансии повторов наследуется почти исключительно по мужской линии [Telenius Н. et al., 1993; Kremer В. et al., 1995; Ranen N. et al., 1995; Nance M., 1996]. Таким образом, генетическая нестабильность мутантного аллеля реализуется преимущественно в мужском гаметогепезе, что и обусловливает «эффект отцовской передачи», характерный для хореи Геитингтопа. Нарастание длины повтора при передаче мутантного гена в ряду поколений закономерно сопровождаетея более ранней манифестацией симптомов болезни у потомков (в наших наблюдениях в среднем на 4-10 лет). является молекулярной основой антиципации в семьях, отягощенных хореей Гентингтона.
По сравнению с хореей Гентингтона другие виды наследственных хореических гиперкинезов являются значительно более редкими. Генерализованная хорея с аутосомно-доминадтиым типом наследования может быть одним из атипичных фенотипов дентаторубро-цал- лидолюисовой атрофии (ДРПЛА), обусловленной экспансией CAG-повторов в гене па хромосоме 12р (фенотип «псевдохореи»). Характеристика ДРПЛА и подходы к прямой ДНК-диагностике данного нейродегенера- тивного заболевания подробно рассматриваются в разделе 3.3.1. Ген наследственной доброкачественной хореи- редкого аутосомно-доминаптного заболевания, характеризующегося появлением гиперкинезов в детском возрасте, непрогрессирующим течением и отсутствием когнитивных нарушений, локализован на длинном плече 14-й хромосомы. На хромосоме 9q21 картирован ген хореи-акаихопитоза (нейроакантоцитоза) - аутосомно- рецессивного заболевания, проявляющегося сочетанием мультисистемных неврологических проявлений (хорея, атаксия, деменция, амиотрофии и др.) с характерными «звездчатыми» изменениями мембран эритроцитов.
В соответствующих информативных семьях возможно проведение косвенной ДНК-диагностики наследственной доброкачественной хореи и хореи-акантоцитоза с использованием полиморфных маркеров сцепленных с локусами 14q и 9ql3. Молекулярная диагностика (и в первую очередь анализ тринуклеотидных CAG-повторов в гене гентингтина) является ключевым исследованием при дифференцировании наследственных хореических синдромов с различными фенокопиями - ревматической (малой) хореей, церебральными васкулитами при некоторых специфических инфекциях и системных заболеваниях соединительной ткани, сенильной хореей и т.д. В качестве примера приводим следующее клиническое наблюдение.
Больной М.Н., 73 лет, заболел остро: на следующий день после прививки от дифтерии появились насильственные движения в руках, ногах, туловище, изменилась походка. После стационарного обследования по месту жительства направлен в НИИ неврологии РАМН с подозрением на хорею Гентинггтона. Семейный анамнез не отягощен. При поступлении в неврологическом, статусе: распространенный крупноразмаилистыи хореический гиперкинез в конечностях, туловище; «танцующая» походка; отмечается также ритмичный тремор пальцев вытянутых рук и тремор покоя в дистальных отделах рук; со стороны координации, рефлекторной и интеллекту аль- но-мнестической сферы существенных нарушении не отмечено. На протяжении месяца указанные гиперкинезы постепенно регрессировали. При компьютерной томографии головного мозга выявлено симметричное расширение субарахноидального пространства больших полушарий, III и боковых желудочков. Комплексное ультразвуковое исследование сосудов мозга (включая дуплексное сканирование), грубое двустороннее распространенное стенозирующес
атеросклеротическое поражение магистральных артерии головы. Результаты прямой ДНК-диагностики (исследование гена гентингтина): выявлены 2 аллеля с нормальным числом, копий тандемных CAG-повторов (менее 30 повторов). Диагноз хореи Геитингтона исключен. Принимая во внимание характер течения заболевания, особенности клинического синдрома и данные лабораторно-инструментальных исследований, состояние больного расценено как постпрививочный инфекционно-аллергический васкулит сосудов головного мозга на фоне вырао/сенного церебрального атеросклероза.
Следует подчеркнуть, что в настоящее время в развитых странах мира в связи с большими успехами в лечении ревматизма и других системной заболеваний соединительной ткани хорея Геитингтона (включая ее атипичные варианты) является основной причиной развития генерализованного хореического гиперкинеза. 11оэтому во всех подобных случаях должно предусматриваться прямое ДНК-тестированис с номощыо доступных молекулярно-генетических методов.