
Гепатолентикулярная дегенерация, или болезнь Вильсона-Коновалова - тяжелое аутосомно-рецессив- ное наследственное заболевание нервной системы и внутренних органов, характеризующееся генетически обусловленным нарушением метаболизма меди и избыточным ее отложением в различных органах и тканях (в первую очередь - в мозге, печени и почках) [Иванова- Смоленская И.А. и др., 1998 (a); Scheinberg I., Stemlieb I., 1984]. Без лечения заболевание имеет неуклонно прогрессирующее течение, с неизбежным: летальным исходом спустя 5-10 лет от момента появления первых симптомов. Гепатолентикулярная дегенерация отличается значительным полиморфизмом неврологических и соматических проявлений, что нашло свое отражение в существовании различных классификаций клинических форм болезни.
В нашей стране общепринятой является подразделение гепатолентикулярной дегенерации на 5 основных форм, предложенное в 1960 году классиком отечественной неврологии Николаем Васильевичем Коноваловым. В соответствии с классификацией Н.В. Коновалова, выделяются следующие формы болезни:
- брюшная форма (преневрологическая) - манифестирует в возрасте от 5 до 17 лет и характеризуется различными вариантами поражения печени, нередко принимающими злокачественное «галопирующее» течение;
- аритмогиперкинетическая форма - возникает в раннем возрасте и проявляется полиморфными инва- лидизирующими экстрапирамидными гиперкиисзами (чаще торсионно-дистонического характера), интеллектуальным снижением и тяжелыми висцеральными расстройствами;
- дрожательно-ригидная форма - возникает в возрасте от 15 до 25 лет и проявляется различным соотношением ригидности, статокинетического тремора, психических и висцеральных проявлений;
- дрожательная форма - характеризуется поздним началом (20-30 лет), наиболее доброкачественным течением и постепенным нарастанием характерного дрожательного гиперкинеза, длительной сохранностью интеллекта;
- экстрапирамидно-корковая форма - представляет собой финал перечисленных выше форм и характеризуется присоединением к типичным двигательным расстройствам пирамидных парезов и очаговых эпилептических пароксизмов. Под нашим наблюдением в ней- рогенетичсском отделении НИИ неврологии РАМН на протяжении 40-летнего периода находились свыше 550 больных гепатолеитикулярной дегенерацией, что позво- 1шло подтвердить обоснованность вышеуказанной систематизации клинических проявлений болезни и детально изучить особенности течении отдельных ее форм [Ивапова-Смол#нская И.А. и др., 1998 (а)].
Клиническая диагностика гепатолеитикулярной дегенерации базируется на выявлении таких характерных изменений медно-белкового обмена, как роговичное кольцо Кайзера-Флейшсра, снижение концентрации в сыворотке крови медь-содержащсго белка церулоплазмина, гиперэкскреция меди с мочой, повышение концентрации свободной фракции меди и снижение концентрации связанной фракции меди в сыворотке крови, повышение содержания меди в биоптате печени.
Гепатолентикулярная дегенерация являет собой редкий пример наследственного заболевания, для которого благодаря знанию основного патогенетического фактора еще до идентификации гена были разработаны высокоэффективные методы лечения. Современная патогенетическая терапия гепатолентикулярной дегенерации основана на использовании медь-элиминирующих препаратов - главным образом, D-пеницилламина, три- ентина и солей цинка [Тимербаева С.Л., 1991; Иванова- Смоленская И. А. и др., 1998 (a); Scheinberg]., Stembeb I., 1984]. D-пеницилламин и триентин - хелатные комп- лексоны, образующие с медью прочные высоко'аффин- ные соединения, которые легко выводятся из организма с мочой. Действие ионов цинка основано на конкурентном ингибировании металлотеининов, что затрудняет всасывает меди в кишечнике и вытесняет ее из белковых тканевых депо. В настоящее время своевременно начатая терапия гепатолентикулярной дегенерации либо предотвращает появление клинических признаков заболевания (превентивное лечение), либо ведет к их полному или частичному регрессу, восстанавливая трудоспособность и существенно улучшая состояние больных [Лекарь П.Г., Макарова В.А., 1984; Иванова-Смоленская И.А. и др., 1998 (а)]. В связи с возможностью эффективного патогенетического лечения ключевым при гепатолентикулярной дегенерации является вопрос адекватной и максимально более ранней диагностики болезни. Между тем до настоящего времени общепринятая лабораторно-инструментальная диагностика болезни остается весьма непростой, а в 5% случаев результаты исследования обмена меди бывают сомнительными, особенно в начальной стадии болезни [Шайнберг Г., 1996]. Таким образом, весьма актуальным и жизненно необходимым становится внедрение в практику надежных методов ДНК-диагностики гепатолентикулярной дегенерации.
Ген гепатолентикулярной дегенерации локализован на длинном плече 13-й хромосомы (локус 13ql4.3) [Frydman М. et al., 1985]. Картирование гена позволило разработать методы косвенной ДНК-диагностики болезни, в том числе на нресимптоматической и пренатальной стадии. В России впервые успешная косвенная ДНК- диагностика гепатолентикулярной дегенерации была проведена сотрудниками нейрогенетического отделения НИИ неврологии РАМН совместно с лабораторией ДНК- диагностики Медико-генетического научного центра РАМН [Воронцова Н.И. и др., 1996]. Ген болезни, получивший обозначение АТР7В, идентифицирован независимыми исследовательскими группами из США и Ка- надыв 1993 году [Bull P.et al., 1993;TanziR. etal., 1993]. Ген ATP7B содержит 22 экзона и кодирует синтез медь- транспортирующей АТФ-азы P-типа. Данный белок в норме обеспечивает встраивание меди в молекулу апо- церулоплазмина в аппарате Гольджи и экскрецию с желчью избытка меди в составе церулоплазмина и других медь-содержащих белков клетками печени [Сох D., 1995; Chowi imootoo G. et al., 1996]. Результатом нарушения функции АТР7В у больных гепатолентикулярной дегенерацией является токсическое накопление меди в печени, а в последующем и в других тканевых депо.
После открытия первичного молекулярного дефекта появилась реальная возможность осуществлять непосредственный анализ повреждений в гене, ведущих к развитию гепатолентикулярной дегенерации, сопоставлять характер мутаций в различных популяциях, а также внедрить прямые методы ДНК-диагностики данного заболевания. К настоящему времени у больных ге- патолентикулярной дегенерацией из большого числа популяций мира идентифицировано уже более 100 различных мутаций в гене АТР7В, в основном представляющих собой нуклеотидные замены и короткие делеции/ вставки [BullP. etal., 1993; Tanzi R. etal., 1993; Thomas G. et al., 1995; Shah A. et ah, 1997]. Большинство больных являются компаунд-гетерозиготами, т.е. имеют различные мутации АТР7В на каждой из двух мутантных хромосом. В разных этнических группах спектр мутаций гена АТР7В у больных имеет свои особенности. Наиболее частой мутацией является замена С—»А в 1069-м кодоне гена (экзон 14), ведущая к замещению аминокислоты гистидин на глютамин. Указанная мутация Hisl069Gln встречается в большинстве европейских и северо-американских популяций (в среднем 20-30% всех мутантных хромосом) [Tanzi R. et al., 1993; Thomas G. et al., 1995; Shah A. et al., 1997]. Кодон 1069 гена ATP7B представляет собой «горячую точку» мутаций, поскольку замена Hisl069Gln ассоциирована с разными гапло- типами [Tanzi R. et al., 1993] и, следовательно, возникает неоднократно у различных больных в результате независимых повторных мутационных событий на разных хромосомах Другие относительно частые кластеры мутаций, характерные для определенных популяций мира, выявляются преимущественно во 2-м, 3-м, 8-м, 1 5-м, 16-м и 18-м экзонах гена [Bull Р. et al., 1993; Thomas G. et al., 1995; Shah A. et al., 1997].
В связи с большим размером гена АТР7В и разнообразием мутаций в нем прямая ДНК-диагностика гепатолентикулярной дегенерации представляет собой непростую задачу. Стандартный подход основан на SSCP- анализе отдельных экзоиов гена с последующим прямым секвенированием образцов, имеющих аномальную электрофоретическую картину. Существенно упрощает прямую ДНК-диагностику болезни установление характерного спектра мутаций АТР7В в конкретной изучаемой популяции и выделение мажорных мутаций, встречающихся в данной популяции с высокой частотой. Обнаружение таких мажорных мутаций позволяет проводить ДНК-диагностику в данном регионе и/или этнической группе, избегая необходимости исследовать всю кодирующую область гена. Указанный подход был реализован нами при проведении молекулярно-генетического анализа гепатолентикулярной дегенерации в славянских семьях, проживающих в европейской части России [Ка-
у
рабанов А.В. и др., 1998; Ivanova-Smolenskaya I. et al., 1999] Нами были обследованы 42 больных гепатолентикулярной дегенерацией, не связанных кровным родством и поступивших в нейрогенетическое отделение НИИ неврологии РАМН в 1992-1998 гг. Мажорная мутация Hisl069Gln (14-й экзон гена) бгдпа обнаружена в обследованной группе почти на половине мутантных хромосом (рис. 56). Аналогичные результаты о преобладании Hisl069Gln (73% хромосом) были получены в этнически близкой польской популяции [Czlonkowska А.,
- ; интересно отметить также, что в недавно проведенном исследовании замена Hisl069Gln выявлена на 43% мутантных хромосом в популяции больных гепато- ^геитикулярной дегенерацией в Башкирии [Карунас А.С.,
- .
По нашим данным, суммарно данная мутация выявляется в гомозиготном состоянии или в компаунде с другими мутациями более чем у 60% больиых-славяп в европейской части России, что позволяет считать выяв- пение данной нуклеотидной замены важнейшим диагностическим тестом при обследовании больных в ука-
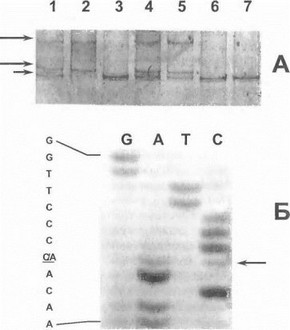
Рис. 56. Прямая ДНК-диагностика мажорной мутации Hisl069Gln в гене АТР7В у больных гепатолентикулярной дегенерацией
А. SSCP-анализ 14-го экзонатена. Дорожка 2 - гомозиговость по нормальному аллелю, дорожки 1,4,5 -тетерозт оты по нормальному аллелю и мутации, дорожки 3,6,7 - гомозиготы по мутантному аллелю. Длинные стрелки указывают на продукты амплификации нормального аллеля, короткая стрелка—на аномальнывелект рофоретический фрагмент (мутация). Б. Прямое секвенирование 14- го экзона. Стрелкой обозначена область мутации (нуклеотидная замена С—gt;А в 1069-м кодоне).
занной этнической группе. В настоящее время в нашей клинике скрининг на мутацию Hisl069Gln в 14-м экзо- не гена АТР7В является неотъемлемой частью диагностического алгоритма у больных гепатолентикулярной дегенерацией.
В нашей работе были исследованы также некоторые другие экзоны гена, которые, согласно литературным данным, наиболее часто повреждаются у больных гепатолентикулярной дегенерацией (экзоны 15,16 и 18); проведенный анализ показал отсутствие мутаций в данных областях АТР7В, за исключением делении с1е13400С в 15-м экзоне, выявленной нами в нескольких семьях. Можно заключить, что спектр мутаций у больных славянского этнического происхождения в нашей стране существенно отличается от большинства других цзучен- I [ых популяций мира.
Большое число повреждений в гене АТР7В и раз- I гообразие возможных комбинаций мутаций у компаунд- гетерозигот, несомненно, являются основными причинами выраженного фенотипического полихгорфизмад»- 11атолентикулярной дегенерации. Анализ больших серий случаев гепатолентикулярной дегенерации, проведенный различными авторами, позволил установить основные закономерности корреляций между характером мутаций в гене АТР7В и особенностями клинической картины заболевания. Наиболее неблагоприятное течение болезни с ранним началом и тяжелыми соматическими проявлениями (фульминантный «вильсоновский» гепатит и Др.) обычно характерно для нонсенс-мутаций, приводящих к преждевременному обрыву трансляции и отсутствию белкового продукта с мутантной копии гена | Thomas G. et al., 1995]. Миссенс-мутации в болынин- с тле случаев характеризуются более мягким течением и развитием полиморфных неврологических и соматических проявлений гепатолентикулярной дегенерации, что обусловлено наличием у синтезируемого мутантного белка частичной функциональной активности [Thomas G. et al., 1995; Shah A. et al., 1997.]. Следует отметить, что некоторые миссенс-мутации, затрагивающие функционально важные участки белка АТР7В, также приводят к тяжелым фенотипическим проявлениям [Shah A. et al., 1997]. В наших исследованиях было установлено, что носительство мутации Hisl 069Gln (особенно на обеих мутантных хромосомах) характеризуется достаточно тяжелым течением болезни и развитием ряда осложнений в ответ на назначение D-пеницилламина (таких как выраженная тромбоцитопения) [Ivanova- Smolenskaya I. et al., 1999]. Наш вывод о неблагоприятном течении болезни у носителей мутации Hisl069Gln, подтверждаемый результатами дискриминантного анализа (рис. 57), совпадает с мнением Shah А. с соавторами (1997), но противоречит данным других исследователей [Bull Р. et al., 1993; Thomas G. et al., 1995]. По- видимому, фенотипическая экспрессия дайной мутации определяется совокупностью генетических и иных факторов, специфичных для каждой конкретной популяции (например, наличие модифицирующих генов, уровень инбредности, характер питания, природио-средовые факторы и т.д.) [Thomas G. et al., 1995; Shah A. et al., 1997]. Указанные наблюдения имеют важное практическое значение и демонстрируют необходимость принимать во внимание характер выявленных у больного мутаций в гене АТР7В при решении вопросов лечения и определении прогноза болезни.
Иллюстрацией роли прямой ДНК-диагностики гепатолентикулярной дегенерации на пре симптоматической стадии болезни является следующее наше наблюдение.
Больная М-ц Т, 26 лет, находилась на обследовании и лечении в нейрогенетическом отделении НИН
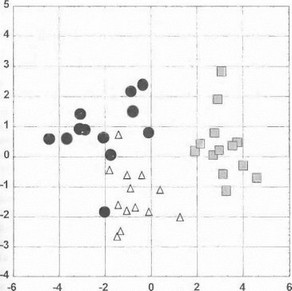
Рис. 57. Результаты дискриминантного анализа у больных гепатолентикулярпой дегенерацией
Показано расположение наблюдений грех i цупп больных в многомерном пространстве признаков (основных клинико-биохимических показателей) в проекции на плоскость канонических координат. Квадратные символы — гомозиготные носители мутации Hisl069Gln; треугольные символы - больные, имеющие мутацию !Iisl069Gln в компаунде с другой мутацией; круглые символы - носители других мутаций в гене АТР7В. На рисунке видно, что наблюдения разных групп формируют относительно компактные кластеры, причем гомозиготы по мутации ] lis 1069GIn являются наиболее изолированными, что свидетельствует об их существенных ыличиях по анализируемым клинико-биохимическим параметрам СП двух других групп.
неврологии РАМН с 31.03 по 30.04.97 г. Больна с 1990 года, когда в возрасте 19 лет впервые появились носовые кровотечения. Спустя 2 года присоединилось и стало нарастать дрожание рук и головы. В декабре 1996 году нейроофталъмологом выявлено кольцо Кайзера- Флепшера, диагностирована гепатолентикулярная де- генерация (дрожательная форма). При поступлении: состояние удовлетворительное, существенных изменений со стороны внутренних органов нет. В неврологическом статусе: гипомимия; скандированная речь; тремор головы по типу «нет-нет»; круппоразмаилистый «порхающий» тремор рук с выраженным иитенциоииым компонентом; со стороны мышечного тонуса, рефлекторной сферы - без особенностей; пошатывание при ходьбе. В общем анализе крови отмечается снижение содержания тромбоцитов до 120 ООО. В сыворотке крови снижено содержание церулоплазмина -12 мг% (в норме 25-45 мг%) и общей меди —4,3 мкмоль/л (в норме 12-24 мкмоль/л). При компьютерной томографии головного мозга патологических изменений плотности вещества мозга не обнаружено; отмечается слабое расширение 3-го и боковых желудочков, слабое расширение охватывающей цистерны, сильвиевых щелей, субарахноидального пространства больших полушарий. Ультразвуковое исследование органов брюшной полости: печень не увеличена в размерах, отмечается диффузная неоднородность ткани печени; эхоструктура паренхимы селезенки однородная, контуры четкие, ровные, размеры не увеличены.
При молекулярно-генетическом исследовании была выявлена мутация Hisl069Gln в гомозиготном состоянии, что подтвердило ранее поставленный диагноз гепатолентикулярной дегенерации.
В отделении обследована младшая сестра больной. У консультируемой 18-летней девушки в течение последнего года отмечались повторные кровотечения из носа при отсутствии каких-либо неврологических симптомов. Кольцо Кайзера—Флеишера отсутствовало, уровень церулоплазмина в сыворотке крови соста вил 18,5 мг%, что не позволяло с достоверностью судить о ее клиническом статусе (норма 25-45 мг%). Данные других лабораторно-инструментальных методов обследования (за исключением сниженного количества тромбоцитов в крови 108 ООО) в пределах нормы. При проведении ДНК-диагностики было выявлено, что консультируемая (как и ее больная сестра) является гомозиготным носителем щтации IBs 1 Об9Gln в гене АТР7В. Девушке был поставлен диагноз «дог/еврологи- ческая стадия гепатолеитикулярной дегенерации» и начато лечение солями цинка по стандартной схеме. Больная продою/сает находиться под наблюдением, на фоне постоянном) лечения неврологической симптоматики не отмечается.
Таким образом, в данном наблюдении идентификация мутации в гене АТР7В оказалась решающим Диагностическим тестом при проведении обследования больной.
Подученный нами опыт прямой Д11К-д и агностики заболевания должен лечь в основу системы мсдико- Генетического консультирования и профилактики гепа- толентикуляриой дегенерации в нашей стране.