
Фактически синдром Ди Джорджи тоже является ТКИН, хотя формально в эту группу его не включают. Заболевание обусловлено утратой гена Tbxl в результате делеций в хромосоме 22q11, варьирующих по протяженности. Ген Tbxl, кодирует транскрипционный фактор Т-box!, главная функция которого — запуск процесса инвагинации жаберных дуг при закладке тимуса. Но при мутации Tbxl не происходит правильной закладки не только тимуса, но и нервной трубки, крупных сосудов, сердца. Синдром проявляется гипоплазией тимуса, в основе которой лежит дефект развития тимического эпителия, обусловленный нарушением экспрессии фактора Т-boxl в производных 3-го глоточного кармана.
Х-сцепленная агаммаглобулинемия Брутона
Гуморальный иммунодефицит, обусловленный мутацией гена BTK, локализованного в Х-хромосоме и кодирующего тирозинкиназу btk (Bruton’s tyrosine kinase) (рис. 4.44). Эта киназа принадлежит к семейству Src и связана с Iga/p-корецепторами комплекса BCR. Btk играет важную роль в проведении активационного сигнала не только от зрелого BCR, но и от проторецептора пре-BCR (см. раздел 3.3.1.2). В развитии Х-сцепленной агаммаглобулинемии имеет значение нарушение передачи сигнала от пре-BCR, определяющее блок развития В-лимфоцитов на стадии про-В-II. В результате В-лимфо- циты практически отсутствуют или содержатся в лимфоидных органах в очень малых количествах, концентрация сывороточных иммуноглобулинов снижена (IgG lt;2 мг/мл, IgA lt;0,2 мг/мл), иммунная защита, обусловленная гуморальным иммунитетом, дефектна. Страдает преимущественно защита от внеклеточных патогенов.
Клинически Х-сцепленная агаммаглобулинемия начинает проявляться с 5—6 мес жизни ребенка, когда практически исчезают материнские иммуноглобулины. У большинства пациентов развиваются хронические рецидивирующие инфекционные процессы, наиболее часто вызываемые Streptococcus pneumoniae и Haemophilus influenzae. Эти бактерии вызывают пневмонии,
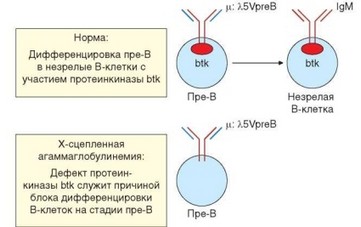
Рис. 4.44. Молекулярный механизм Х-сцепленной агаммаглобулинемии Бруттона. Дефицит протеинкиназы btk, вызванный мутацией, блокирует созревание пре-В-клеток в зрелые В-лимфоциты
отиты, синуситы, конъюнктивиты. Для больных с Х-сцепленной агамма- глобулинемией характерна повышенная чувствительность к микоплазмам и уреаплазмам, вызывающим хронические пневмонии, артриты, цистит, инфекционное поражение подкожной клетчатки. У больных часто развиваются Th1-зависимые аутоиммунные процессы — ревматоидный артрит, склеродермоподобный синдром, неспецифический язвенный колит и др. Нередко выявляют деформацию грудной клетки, вызванную хроническим бронхо-легочным процессом.
runep-IgM-синдром (Hyper IgM-syndrome — HIGM)
Гетерогенный первичный иммунодефицит, в основе которого лежат мутации, сцепленные и не сцепленные с полом. К Х-сцепленным вариантам относят гипер-IgM-синдром 1 (HIGM1), в основе которого лежит дефект рецептора Т-клеток CD40L (CD154) (рис. 4.45), и ангидротическая эктодермальная дисплазия, возникающая в результате мутации в сигнальной молекуле IKKy. Описано 3 варианта аутосомно-рецессивных форм гипер- IgM-синдрома: HIGM2, HIGM3 и HIGM4. Гипер-^М-синдром 2 (HIGM2) связан с мутацией в гене, кодирующем активационно-индуцируемую цити- диндезаминазу (AID), участвующую в переключении изотипов Н-цепей и ответственную за гипермутагенез, который обеспечивает повышение аффинтета антител. У больных с мутацией AID повышен уровень IgM и снижен уровень IgG при нормальной экспрессии CD40L. Гипер-^М-синд- ром 3 (HIGM3) связан с мутацией в гене, кодирующем молекулу CD40. Эта мутация приводит к тем же последствиям, что и мутация CD40L. Сходная симптоматика обнаружена при мутациях гена UNG, кодирующего фермент урацил-К-гликозилазу, участвующего в переключении классов иммуноглобулинов в В-клетках.
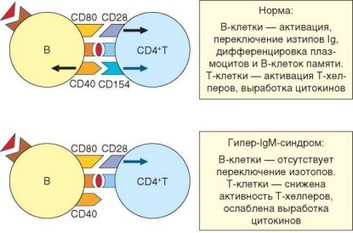
Рис. 4.45. Молекулярная основа ranep-IgM-синдрома. Клинические проявления ranep-IgM-синдрома развиваются при дефекте не только CD40L (CD154), но и CD40, а также некоторых связанных с ними сигнальных факторов
Патогенетическая основа всех перечисленных форм гипер-IgM-синд- рома — нарушение процесса переключения классов иммуноглобулинов, кодируемых С-генами Н-цепей. Чаще всего основа дефекта — отсутствие или неполноценность взаимодействия молекулы CD40 (костимулирующая молекула В-лимфоцитов, макрофагов и дендритных клеток) с ее лигандом — CD154 (активационная молекула Т-лимфоцитов; см. раздел 3.4.2.2). Именно дефект лиганда служит наиболее частой причиной дефекта переключения классов иммуноглобулинов (гипер-IgM-синдром 1). Однако роль указанного взаимодействия не ограничивается переключением изотипов. Сигналы, порождаемые взаимодействием, обеспечивают активацию всех АПК, на которых экспрессируется CD40. Это взаимодействие необходимо для индукции пролиферации В-клеток и экспрессии костимулирующей молекулы CD80 на их поверхности. Через молекулу CD40 передаются активирующие сигналы в макрофаг и дендритную клетку, что повышает их антигенпрезентирующую и другие формы активности. Обусловленное этим ослабление активности АПК сказывается на эффективности презентации антигена и определяет ослабление функции Т-лимфоцитов, прежде всего Th1-клеток.
Наиболее очевидный признак гипер-IgM-синдрома — нарушение баланса классов иммуноглобулинов — снижение концентарции IgG и IgA при повышенном или нормальном содержании IgM. В подавляющем большинстве случаев патология развивается у мальчиков (в соответствии с преобладанием Х-сцепленного варинта гипер-IgM-синдрома 1). Основу клинической картины составляют тяжелые рецидивирующие инфекционные заболевания на 1-м году жизни. Чаще всего выявляют заболевания,
вызываемые внутриклеточными патогенами: пневмоцистную пневмонию; диарею, вызываемую Cryptosporidium; склерозирующий холангит; парвови- рус-индуцированную апластическую анемию.
Общая вариабельная иммунная недостаточность
Это группа заболеваний с различным патогенезом, выясненным не для всех вариантов. В большинстве случаев мутации затрагивают гены, которые кодируют белки, вовлеченные в В-лимфопоэз и гомеостаз В-клеток. Чаще других выявляют мутации двух белков этой группы — рецептора гомеостатического фактора В-клеток BAFF (фактор выживания В-клеток; относится к семейству TNF) и Blimp1 — белка, индуцирующего дифференцировку плазматических клеток (см. раздел 3.6.2.3). При заболеваниях этой группы преобладают дефекты гуморального иммунитета со снижением концентрации иммуноглобулинов. Однако выявляют нарушения и в Т-клеточном звене — снижение содержания NK- и CD4+ Т-клеток с усилением экспрессии маркеров клеток памяти — CD45R0 и CD29. Нарушение экспрессии и функционирования ряда мембранных молекул Т-клеток приводит к ослаблению их костимуляции при презентации антигена и, вследствие этого, нарушению секреции IL-2 и пролиферативной экспансии клонов Т-лимфоцитов. Показана ориентация антигензависимой дифференцировки Т-клеток больных в направлении Thl-клеток (одна из причин — усиление выработки IL-12), что обусловливает ослабление гуморального иммунного ответа.
Общий вариабельный иммунодефицит часто диагностируют клинически в возрасте 20—40 лет. Наиболее частые проявления синдрома — повторные пневмонии, синуситы, артрит, тромбоцитопеническая пурпура, аутоиммунная гемолитическая анемия, колит. Инфекционные поражения чаще затрагивают респираторный и желудочно-кишечный тракты и вызываются разнообразными бактериями, вирусами, а также грибами и паразитами. У больных повышена частота развития злокачественных опухолей.
Селективный IgA-дефицит
Самый распространенный из первичных иммунодефицитов (частота 1 на 500—1000). Его основа — сниженная концентрация сывороточного и секреторного IgA. Клинически проявляется только в 50% случаев — в виде рекуррентных инфекций. Иногда сочетается с дефицитом IgG2; в этом случае выявляют более тяжелое течение заболевания. Молекулярный механизм заболевания неизвестен. В развитие заболевания вовлечен функциональный дефицит TGFpi, принимающий участие в переключении генов иммуноглобулинов на IgA и, возможно, IgG2.
Гипер-IgE-синдром (синдром Иова)
Характеризуется высоким уровнем сывороточного IgE (gt;2000 МЕ/мл) и эозинофилией. Для него свойственны повторные абсцессы кожи и подкожной клетчатки при отсутствии местной гиперемии, гипертермии, болевого синдрома. Характерно развитие пневмоний с формированием пневмоцеле. Молекулярно-генетическая природа не установлена.
Сидром Вискотта—Олдрича
Х-сцепленный иммунодефицит, обусловленный мутацией гена WASP, локализованного в Х-хромоме. Это ген кодирует белок WASP (Wiscott-Aldrich syndrome protein), играющий важную роль в функционировании цитоскелета. Он регулирует полимеризацию актина. Нормальная функция этого белка необходима для полноценной подвижности клеток, их поляризации, формирования филоподий при хемотаксисе, адгезии клеток и образования иммунного синапса при взаимодействии клеток иммунной системы.
В зависимости от локализации мутаций и протяженности затронутого ими участка гена развивается три клинических варианта заболевания: полномасштабный синдром Вискотта-Олдрича (следствие делеций) и варианты с изолированным проявлением тромбоцитопении или нейтропении. Классическая картина синдрома Вискотта-Олдрича характеризуется тром- боцитопенией с образованием тромбоцитов малого размера, экземой и рекуррентными инфекциями.
Для синдрома Вискотта-Олдрича характерны множественные нарушения в иммунной системе, затрагивающие преимущественно фагоцитарную и цитолитическую активность клеток врожденного иммунитета, т.е. функции, наиболее зависящие от движения клеток и активного участия цитоскелета. Нарушение образования иммунного синапса между Т-лимфоцита- ми и АПК влияет на все проявления адаптивного иммунитета.
Атаксия-телеангиоэктазия (синдром Луи-Бар)
Наследственное заболевание, обусловленное дефектом гена АТМ (Ataxia telangiectasia mutated). Относится к заболеваниям, в основе которых лежит синдром хромосомных поломок. Заболевание развивается вследствие мутаций, возникающих в любых участках гена АТМ. Результатом мутаций может быть полное отсутствие или ослабление синтеза белка АТМ, а также синтез функционально неполноценного белка.
Белок АТМ — серинтреониновая протеинкиназа. Его основная функция состоит в инициации сигналов репарации разрывов двуцепочечной ДНК, возникающих как в физиологических условиях (при мейозе, перестройке V-генов антигенраспознающих рецепторов и т.д.), так и индуцируемых действием внешних факторов (например, ионизирующей радиации). При разрывах ДНК АТМ-киназа аутофосфорилируется и переходит из димерной в мономерную форму. АТМ-киназа обеспечивает фосфорилирование белков комплекса MRN и связанных с ним факторов, непосредственно осуществляющих репарацию ДНК. В случае небольшого числа разрывов они успешно выполняют эту функцию. При невозможности успешной репарации развивается апоптоз, запускаемый с участием фактора р53. Отсутствие полноценной репарации ДНК обусловливает нестабильность генома, следствием чего является повышение радиочувствительности клеток, частоты развития злокачественных опухолей, особенно лимфом и лейкозов.
Наиболее характерный клинический признак атаксии-телеангиоэктазии — нарастающая атаксия, проявляющаяся изменением походки. Она обусловлена нейродегенерацией с атрофией мозжечка. Развитие нейродегенеративных процессов связано с тем, что при созревании нейронов головного мозга происходят процессы рекомбинации ДНК, сопровождающиеся ее двойными разрывами. Другой симптом, определивший название заболевания, — телеангиоэктазия — представляет устойчивую дилатацию глазных и лицевых кровеносных сосудов.
Нарушение репарации разрывов ДНК, происходящих при созревании Т- и В-лимфоцитов, лежит и в основе иммунодефицита, наблюдаемого при атаксии-телеангиоэктазии. Иммунодефицит проявляется в хронических рецидивирующих бактериальных и вирусных инфекционных заболеваниях бронхолегочного аппарата, что обычно служит причиной смерти больного.
Синдром Ниймегена
Ниймеген — город в Голландии, в котором впервые описан синдром. Это наследственное заболевание относят к синдромам хромосомных поломок, сопровождающихся формированием нестабильности генома. Развитие этого заболевания связано с мутацией в гене NBS1, продукт которого — нибрин — участвует в репарации ДНК в составе комплекса MRN, являясь субстратом для фосфорилирования протеинкиназой АТМ. В связи с этим и патогенез, и клинические проявления синдрома Ниймегена практически совпадают с таковыми при атаксии-телеангиоэктазии. В обоих случаях развиваются нейродегенеративные изменения, однако при синдроме Ниймегена преобладают явления микроцефалии, поскольку процессы рекомбинации ДНК происходят и при созревании нейронов головного мозга.
Аутоиммунный лимфопролиферативный синдром
Заболевание характеризуется нарушением апоптоза и связанными с этим незлокачественной лимфопролиферацией, гипериммуноглобулинеми- ей, аутоиммунными процессами, увеличением содержания CD3+ CD4- CD8- клеток в крови. Мутации, лежащие в основе синдрома, чаще всего локализуются в гене TFRRSF6, кодирующем Fas-рецептор (CD95). К клиническим проявлениям ведут только мутации, вызывающие изменения во внутриклеточном участке молекулы CD95. Реже мутации затрагивают гены Fas-лиганда и каспаз 8 и 10 (см. раздел 3.4.1.5). Мутации проявляются ослабленной экспрессией молекул, кодируемых соответствующим геном, и ослаблением или полным отсутствием передачи апоптотического сигнала.
Х-сцепленный лимфопролиферативный синдром
Редкий иммунодефицит, характеризуемый извращенным антивирусным, антиопухолевым и иммунным ответом. Возбудитель Х-сцепленного лимфопролиферативного синдрома — вирус Эпштейна—Барр. Вирус проникает в В-клетки через взаимодействие молекулы gp150 оболочки вируса с рецептором CD21 на клеточной мембране. У больных Х-сцепленным лимфопролиферативным синдромом происходит поликлональная активация В-клеток и беспрепятственная репликация вируса.
Инфицирование вирусом Эпштейна—Барр при Х-сцепленном лимфопролиферативном синдроме — результат мутации в гене SH2D1A, кодируе- щем адапторный белок SAP [Signaling lymphocytic activation molecule (SLAM)- associated protein]. S^-домен белка SAP распознает тирозиновый мотив в цитоплазматической части SLAM и ряда других молекул. Процессы, развивающиеся в клетках иммунной системы при активации, опосредованной через SLAM-рецептор, играют ведущую роль в противовирусном иммунитете. SLAM-рецептор экспрессируется на тимоцитах, Т-, В-дендритных клетках, на макрофагах. Экспрессия усиливается при активации клеток. Регуляторное действие белка SAP связано с подавленнием активности тирозинфосфатаз в
отношении SLAM. В отсутствие SAP фосфатаза SH-2 свободно соединяется с рецептором SLAM, дефосфорилирует его и подавляет передачу сигнала. Не происходит активации главных эффекторов противовирусной защиты — Т- и NK-клеток, что приводит к бесконтрольному размножению вируса Эпштейна—Барр. Кроме того, SAP облегчает взаимодействие тирозинкиназы Fyn с рецептором SLAM, что способствует передаче активационного сигнала.
В разнобразных клинических проявленях X-сцепленного лимфопролиферативного синдрома наиболее постоянны молниеносный инфекционный мононуклеоз, доброкачественные и злокачественные лимфопролиферативные нарушения, а также дисгаммаглобулинемия или гипогаммаглобулинемия. Среди локальных поражений превалирует поражение печени, вызываемое инфильтрацией В-клетками, инфицированными вирусом Эпштейна—Барр, и активированными Т-клетками, которая приводит к некрозу ткани печени. Печеночная недостаточность — одна из главных причин смертности больных X-сцепленным лимфопролиферативным синдромом.
IPEX- синдром
Сцепленный с Х-хромосомой синдром дисрегуляции иммунитета, полиэндокринопатии и энтеропатии (Immune disregulation, polyendocrinopathy, enteropathy X-linked syndrome) развивается как следствие мутаций гена FOXP3, локализованного в Х-хромосоме. FOXP3 является «мастер-геном», ответственным за развитие регуляторных Т-клеток фенотипа CD4+ CD25+. Эти клетки играют центральную роль в сдерживании активности аутоспецифических клонов Т-лимфоцитов на периферии. Дефект гена FOXP3 сопряжен с отсутствием или дефицитом этих клеток и растормаживанием разнообразных аутоиммунных, а также аллергических процессов.
IPEX-синдром проявляется в развитии множественного аутоиммунного поражения эндокринных органов, пищеварительного тракта и половой системы. Это заболевание начинается в раннем возрасте и характеризуется поражением ряда эндокринных органов (сахарный диабет типа I, тирои- дит) с высоким уровнем аутоантител, тяжелой энтеропатией, кахексией, низкорослостью, аллергическими проявлениями (экзема, пищевая аллергия, эозинофилия, увеличение уровня IgE), а также гематологическими изменениями (гемолитическая анемия, тромбоцитопения). Больные дети (мальчики) погибают в течение первого года жизни от повторных тяжелых инфекционных заболеваний.
APECED-синдром
Аутоиммунная полиэндокринопатия, кандидоз, эктодермальная дистрофия (Autoimmune polyendocrinopathy, candidiasis, ectodermal dystrophy) — аутоиммунный синдром, обусловленный дефектом отрицательной селекции тимо- цитов. Его причина — мутации гена AIRE, ответственного за эктопическую экспрессию органоспецифических белков в эпителиальных и дендритных клетках мозговой зоны тимуса, ответственных за отрицательную селекцию (см. раздел 3.2.3.4). Аутоиммунный процесс поражает преимущественно паращитовидные железы и надпочечники, а также островки поджелудочной железы (развивается диабет типа I), щитовидную железу, половые органы.
Часто сопровождается развитием кандидоза. Выявляют также дефекты морфогенеза производных эктодермы.
При рассмотрении спектра первичных иммунодефицитов обращает на себя внимание отсутствие нозологических единиц, связанных с патологией NK-клеток. К настоящему времени описано немногим более десятка мутаций, затрагивающих функцию этих клеток у отдельных лиц, что позволяет сделать заключение о крайней редкости иммунодефицитов, избирательно затрагивающих NK-клетки.