
В этой главе будет рассмотрена общая схема иммунопатологии, развивающейся при первичных иммунодефицитах. Конкретным первичным иммунодефицитам посвящены разделы 4.7.1.4 и 4.7.1.5.
Отдельно рассмотрим патологию, затрагивающую развитие и функционирование клеток иммунной системы. Обращает на себя внимание, что все иммунодефициты, приводящие к нарушению развития клеток иммунной системы, затрагивают исключительно лимфоидные ростки. Заболеваний, в основе которых лежало бы нарушение дифференцировки миелоидных клеток, по-видимому, не существует. Вероятно, соответствующие генетические дефекты затрагивают более широкий спектр клеток и тканей, а потому приводят к гибели развивающихся эмбрионов.
Первичные иммунодефициты, связанные с нарушением развития лимфоидных клеток, наоборот, очень многочисленны. Спектр первичных иммунодефцитов в незначительной степени отражает стадии нормальной дифференцировки лимфоцитов (некоторые данные, касающиеся поражения Т-линии лимфопоэза отражены на рис. 4.42). Наиболее ранний этап развития лимфоидных клеток, на котором могут происходить их гене-
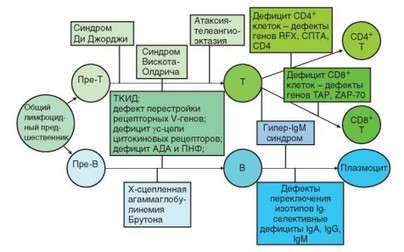
Рис. 4.42. Связь первичных иммунодефицитов с дифференцировкой лимфоцитов. Дефекты развития и функционирования Т-клеток при мутациях могут быть приурочены к определенным этапам их развития
тические изменения, — стадия ранних лимфоидных предшественников. Выживаемость и пролиферацию про-Т- и про-В-клеток обеспечивает IL-7, предшественников NK-клеток — IL-15. Выпадение общей структуры рецепторов этих цитокинов — у(с)-цепи приводит к гибели ранних предшественников T- и NK-лимфоцитов и прекращению развития соответствующих линий лимфоидных клеток, тогда как предшественники В-лимфоцитов выживают за счет избыточности цитокинов, обеспечивающих эту стадию развития В-клеток. Аналогичный эффект в отношении развития Т-клеток достигается при мутациях a-цепи рецептора IL-7 и тирозинкиназы Jak3, связанной с у(с)-цепью. Блокада развития Т-лимфоцитов не дает возможности наблюдать проявления дефектов у(с)-цепи и Jak3 на уровне действия IL-2, IL-4 и других цитокинов, чьи рецепторы содержат у(с)-цепь и передают сигнал с участием Jak3, поскольку клетки не доживают до этих стадий развития.
Другая группа генетических нарушений, влияющих на развитие лимфоцитов, связана с перестройкой генов антигенраспознающих рецепторов и затрагивает Т- и В-лимфоциты. Среди многочисленных факторов, вовлекаемых в перестройку V-генов, в развитии первичных иммунодефицитов участвуют мутации генов рекомбиназ RAG-1 и RAG-2, запускающих эту перестройку, а также ДНК-зависимой протеинкиназы, участвующей в реализации ее промежуточных этапов. Кроме того, на этой стадии проявляется эффект мутации генов АТМ и функционально родственных факторов при атаксии-телеангиоэктазии и других синдромах нестабильности хромосом вследствие неполноценности процессов репарации ДНК. Любые нарушения
реаранжировки V-генов и гибель клеток на этой стадии приводит к блоку развития лимфоцитов, несущих антигенраспознающие рецепторы, т.е. Т- и В-клеток. К аналогичным результатам приводит дефект сборки молекул, связанных с антигенраспознающим рецептором, например, дефект цепей комплекса CD3, нарушающий его экспрессию и развитие Т-клеток.
Несколько разновидностей первичных иммунодефицитов связано с нарушением селекции развивающихся лимфоцитов и дифференцировки субпопуляций. Основная разновидность первичных В-клеточных иммунодефицитов — агаммаглобулинемия Бруттона основана на блоке развития В-лимфоцитов вследствие нарушения передачи сигнала от промежуточного пре-ВCR-рецептора, временно присутствующего на поверхности равзвиваю- щихся В-клеток и свидетельствующего о правильно реализованной реаранжировке V-гена Н-цепей. Несколько Т-клеточных дефицитов обусловлено нарушеннием развития одной из основных субпопуляций, вызванного отсутствием экспрессии молекул MHC-I или MHC-II. Причина этого — мутация как самих генов MHC, так и генов, регулирующих их экспрессию (особенно в случае генов группы MHC-II), а также нарушающих подготовку антигенных пептидов к встраиванию в молекулы MHC (нарушение транспорта пептида при дефектах генов ТАР, — причина отсутствия молекул CD8 и соответствующей субпопуляции Т-клеток). Наконец, могут быть нарушены сигнальные пути, обеспечивающие дифференцировку клеток (дефект ZAP-70 препятствует развитию CD8+ Т-клеток).
Дефект факторов эпителиального микроокружения тимуса — причина нескольких вариантов дефицита Т-лимфоцитов (синдром Ди Джорджи, дис- генезия тимуса). Дефект эктопической экспрессии внетимусных органоспецифических антигенов в эпителиальных клетках мозгового слоя тимуса приводит к неполной элиминации аутоспецифических клонов в ходе отрицательной селекции тимоцитов, что служит основой синдрома APECED.
Первичные иммунодефициты, реализуемые на уровне зрелых клеток адаптивного иммунитета, более редки и затрагивают преимущественно В-лимфоциты. Дефекты гомеостатических факторов BAFF и APRIL приводят к неполноценности процессов, обеспечивающих нормальную численность и функциональную активность этих клеток в периферическом отделе иммунной системы. Существует несколько вариантов селективных дефицитов классов иммуноглобулинов. Самый распространенный из них — селективный дефицит IgA, который, однако, не всегда проявляется клинически. Эта группа иммунодефицитов обусловлена дефектами механизма переключения СН-генов. На уровне лимфоцитов реализуется дефект, лежащий в основе гипер-IgM-синдрома, суть которого сводится к нарушению контактных взаимодействий Т-лимфоцитов и АПК, обусловленных молекулами CD40 и CD154 (мутация гена CD154 — наиболее частая причина этой патологии). Нарушения, связанные с дефектами белка цитоскелета при синдроме Вискотта—Олдрича, также проявляются в значительной степени на уровне функционирования клеток адаптивного иммунитета.
Несколько первичных иммунодефицитов обусловлены дефектностью клеток или гуморальных факторов, сдерживающих иммунные процессы. Примерами могут служить IPEX-синдром, при котором нарушается развитие естественных регуляторных Т-клеток, и аутоиммунный лимфопролиферативный синдром, при котором нарушаются механизмы апоптоза, что приводит к дефекту элиминации аутоспецифических клонов. В обоих случаях происходит растормаживание аутоиммунных процессов и усиленная пролиферация лимфоцитов в местах систематического воздействия антигена (кишечник и т.д.).
Генетические дефекты, затрагивающие врожденный иммунитет, проявляются в основном на стадии реализации его эффектов. Нарушения этой группы могут затрагивать ферменты, обусловливающие формирование факторов бактерицидности (например, NADPH-оксидазы), молекулы адгезии, необходимые для миграции клеток (варианты LAD-синдрома), механизмы высвобождения внутриклеточных гранул (синдром Чедиака—Хигаси). Большую группу образуют иммунодефициты, обусловленные дефектом генов, кодирующих компоненты комплемента. В связи с упоминавшейся выше избыточностью следствия этих дефектов редко бывают тяжелыми. С другой стороны, генетически обусловленный дефект ингибитора C1q вызывает тяжелые последствия в виде отека при вовлечении каскада факторов нескольких систем, участвующих в развитии сосудистых реакций при воспалении.