
(табл. 4.19)
Тяжелая комбинированная иммунная недостаточность
Тяжелая комбинированная иммунная недостаточность (ТКИН, Severe combined immune deficiency — SCID). Комбинированные иммунодефициты характеризуются отсутствием Т-клеток и сильными нарушениями адаптивного иммунитета. Для больных ТКИН характерна ранняя клиническая манифестация заболевания, практически в первые недели жизни ребенка. Характерные признаки — упорная диарея, нарушение всасывания в кишечнике, прогрессирующее поражение бронхо-легочного аппарата, гнойные инфекционные заболевания кожи и слизистых оболочек. Лимфоидная ткань недоразвита. Возбудителями инфекционных заболеваний чаще всего являются условно-патогенные бактерии, грибы, вирусы, простейшие. Заболевание может проявиться после вакцинации БЦЖ в виде локальной или генерализованной БЦЖ-инфекции. При оценке иммунного статуса выявляют лимфоцитопению, снижение содержания и функциональной активности Т-клеток, гипогаммаглобулинемию.
Таблица 4.19. Первичные иммунодефициты, затрагивающие систему адаптивного иммунитета
Продолжение табл. 4.19
Продолжение табл. 4.19
Продолжение табл. 4.19
Продолжение табл. 4.19
Окончание табл. 4.19
Различают 16 вариантов ТКИН.
• Х-сцепленная тяжелая комбинированная иммунная недостаточность [дефицит у(с)-цепи]. Вызвана мутацией локализующегося в Х-хромосоме гена у(с)-цепи, общей для рецепторов для IL-2, IL-4, IL-7, IL-9, IL-15 и IL-21 (рис. 4.43). Самый частый вариант ТКИН — 46% от всех ее случаев. При этой форме иммунодефицита теоретически не должны проявлять свою активность все перечисленные цитокины (из-за дефекта их рецепторов). Однако в реальности важно отсутствие функций только одного цитокина — IL-7, в связи с тем, что он обеспечивает выживаемость лимфоидных предшественников и развитие всех ростков лимфопоэза. У мышей, гомозиготных по мутации scid, отсутствуют Т-, В- и NK-клетки. У человека роль IL-7 в развитии В-лимфоцитов заменима, и больные с Х-сцепленной формой ТКИН не имеют Т- и NK-клеток, тогда как В-лимфоциты у них развиваются практически нормально. Развитие Т-клеток блокируется на стадии перехода DN1 в DN2.
нитей ДНК. Формирование двунитевых разрывов происходит как в процессе реаранжировки V-генов, так и при действии повреждающих факторов, например, ионизирующей радиации. Именно поэтому мутации гена ДНК-зависимой протеинкиназы не только нарушают процесс реаранжировки V-генов, но и препятствуют ликвидации последствий действия радиации и других факторов, повреждающих ДНК. Блокада развития Т- и В-клеток происходит на тех же этапах, как при дефиците RAG-1/RAG-2.
• Дефицит пуриннуклеотидфосфорилазы (PNP). Дефицит обусловлен мутацией гена PNP. Фермент пуриннуклеотидфосфорилаза катализирует превращение гуанозина в гуанин, а также инозина и дезоксиинозина в гипоксантин, что предотвращает накопление токсических продуктов метаболизма гуаниновых нуклеотидов — дезоксигуанозина и гуанозина, токсичных только для Т-лимфоцитов. Дефект пуриннуклеотидфос- форилазы частично компенсируется, в связи с чем он проявляется менее тяжело, чем дефицит аденозиндезаминазы.
Описанная группа ТКИН представляет наиболее тяжелые варианты первичных иммунодефицитов. В большинстве случаев больные дети погибают в течение первого года жизни от инфекций. Хотя не во всех случаях поражаются все линии лимфопоэза (неизменно поражение Т-ростка или субпопуляций Т-лимфоцитов) на уровне функционирования иммунной системы, иммунодефицит всегда является комбинированным в связи с зависимостью всех форм иммунного ответа от Т-лимфоцитов.
Тяжелая комбинированная иммунная недостаточность
Тяжелая комбинированная иммунная недостаточность (ТКИН, Severe combined immune deficiency — SCID). Комбинированные иммунодефициты характеризуются отсутствием Т-клеток и сильными нарушениями адаптивного иммунитета. Для больных ТКИН характерна ранняя клиническая манифестация заболевания, практически в первые недели жизни ребенка. Характерные признаки — упорная диарея, нарушение всасывания в кишечнике, прогрессирующее поражение бронхо-легочного аппарата, гнойные инфекционные заболевания кожи и слизистых оболочек. Лимфоидная ткань недоразвита. Возбудителями инфекционных заболеваний чаще всего являются условно-патогенные бактерии, грибы, вирусы, простейшие. Заболевание может проявиться после вакцинации БЦЖ в виде локальной или генерализованной БЦЖ-инфекции. При оценке иммунного статуса выявляют лимфоцитопению, снижение содержания и функциональной активности Т-клеток, гипогаммаглобулинемию.
Таблица 4.19. Первичные иммунодефициты, затрагивающие систему адаптивного иммунитета
|
Название |
Ген (хромосома) |
Природа дефекта |
Иммунологические проявления |
|
Тяжелая комбинированная иммунная недостаточность |
|||
|
Х-сцепленная тяжелая комбинированная иммунная недостаточность (дефицит у (с)-цепи) |
у(с) №) |
Отсутствие у(с)-цепи, общей для рецепторов IL-7, IL-2 и ряда других цитокинов. Гибель развивающихся Т- и NK-клеток при переходе на стадию DN2 |
Формула: Т В+ NK-. Отсутствие всех видов иммунной защиты, связанной с Т- и NK-клетками |
|
Дефицит тирозинкиназы Jak3 |
JAK3 |
Jak3 передает сигнал от у(с). Последствия дефицита те же |
То же |
|
Дефицит а-цепи рецептораIL-7 |
IL7RA |
Нарушение функционирования рецептора для IL-7, ответственного за раннее развитие Т- и NK-клеток |
Формула: Т В+ NK-. Проявления практически те же, что при Х-сцепленной тяжелой комбинированной иммунной недостаточности |
|
Дисгенезия тимуса (синдром Незелоф) |
Не установлены |
Вследствие гистогенетических дефектов нарушено развитие Т-клеток в тимусе |
Изменена структура популяции Т-клеток, уменьшено их число, снижена функция |
Продолжение табл. 4.19
|
Название |
Ген (хромосома) |
Природа дефекта |
Иммунологические проявления |
|
Дефицит RAG-1/ RAG-2 |
RAG1, RAG2 |
Не происходит перестройки V-генов во всех типах рецепторов. Остановка развития В-клеток на стадии про-В, Т-клеток — на стадии DN2 |
Формула: Т В- NK+. Практически не функционирует адаптивный иммунитет |
|
Синдром Оменна |
Не установлены |
Частичный дефект реаранжировки V-генов антигенрас- познающих рецепторов. Частичное нарушение развития и функциональные дефекты лифмоцитов |
Формула: Т В+ NK-. В наибольшей степени страдает развитие Thl-клеток, синтез IFNy и IL-2 |
|
Дефицит ДНК-зависимой пр отеинкиназы |
Не установлены |
Нарушен процесс разрешения «шпилек» при перестроке V-генов. Нарушено формирование всех типов зрелых рецепторных генов |
Формула: Т В- NK+. Отсутствует адаптивный иммунитет |
|
Дефициты CD3y и CD3e |
CD3G, CD3E |
CD3y и CD3s составляют часть комплекса СD3. При их отсутствии нарушается экспрессия рецепторного комплекса TCR—CD3 на мембране и передача активационного сигнала внутрь Т-клетки |
Формула: Т В+ NK+. Снижено число и ослаблена функция Т-клеток |
|
Дефицит молекул MHC-II |
CIITA, RFXNKRFX5, RFXAP |
В отсутствие молекул MHC-II не происходит дифференцировка и селекция CD4+ Т-кле- ток и презентация им антигена. Поскольку Т-хелперам принадлежит ключевая роль в адаптивном иммунитете, нарушается развитие практически всех его ветвей |
Формула: Т (CD4- CD8+) B+ NK+. Нарушены большинство проявлений адаптивного иммунитета |
|
Дефицит CD4 |
CD4 |
Дефект корецептора CD4 нарушает функции Т-хелперов с теми же последствиями, как при дефиците MHC-II |
Формула: Т (CD4- CD8+) B+ NK+. То же |
Продолжение табл. 4.19
|
Название |
Ген (хромосома) |
Природа дефекта |
Иммунологические проявления |
|
Дефицит ТАР |
TAP1, TAP2 (6p) |
ТАР отвечает за транспорт пептидов из цитозоля в эндоплазматический ретикулум, где они встраиваются в молекулу MHC-I. В условиях дефицита ТАР полноценные MHC-I не формируются и зависящие от них CD8+ Т-клетки не развиваются |
Формула: Т (CD4+ CD8-) B+ NK+. Нарушен цитотоксический Т-клеточный ответ |
|
Дефицит ZAP-70 |
ZAP70 |
Тирозинкиназа ZAP-70 участвует в передаче активационных сигналов. Она важна также для развития CD8+ Т-клеток, дефицит которых составляет основу данной патологии |
Формула: Т (CD4- CD8+) B+ NK+. То же |
|
Дефицит CD25 |
IL2RA |
CD25 — a-цепь IL-2R, экспрессируемая на ранних этапах развития Т-клеток, а также на активированных и регуляторных Т-клет- ках. Дефицит CD25 приводит к возникновению дефектов развития Т-клеток, особенно регуляторных, и нарушению активации зрелых Т-лимфоцитов |
Формула; Т± В+ NK+. Снижено содержание Т-клеток и их функция. Аутоиммунные процессы |
|
Дефицит CD45 |
CD45 |
CD45 активирует киназу Lck, участвующую в запуске активационных сигналов в лимфоциты. При дефиците CD45 этот процесс нарушен |
Формула: Т В+ NK+. Снижена функциональная активность Т- и В-клеток |
|
Дефицит адено- зиндезаминазы (ADA) |
ADA (20q) |
Аденозиндезаминаза катализирует превращение аденозина и дезок- сиаденозина в инозин и дезоксиинозин. Накопление аденозина и дезоксиаденозина приводит к гибели тимо- цитов. Формируется дефицит Т-клеток |
Формула : Т В+ NK+. Нарушены все звенья адаптивного иммунитета, зависящие от Т-клеток |
Продолжение табл. 4.19
|
Название |
Ген (хромосома) |
Природа дефекта |
Иммунологические проявления |
|
Дефицит пурин- нуклеотидфосфо- рилазы (PNP) |
PNP (14q) |
Пуриннуклеотид фосфорилаза катализирует превращение гуанозина в гуанин и инозина и дезокси- инозина в гипоксантин. Метаболиты, накапливающиеся при дефиците PNP, токсичны для развивающихся тимо- цитов, но дефект частично компенсируется. Поэтому он выражен слабее, чем при дефиците аденозиндезами- назы |
Формула: Т+ЫК+В+. Последствия дефицита Т-лимфоцитов выражены слабее, чем при дефиците ADA |
|
Другие первичные иммунодефициты |
|||
|
Синдром Ди Джорджи |
TBX1 (22q) |
Дефекты гена TBХ1, кодирующего фактор Т-box 1, ответственный за инвагинацию жаберных дуг при закладке тимуса. Формирующаяся дис- генезия тимуса приводит к нарушению привлечения в орган клеток-предшест- венников. Развития Т-лимфоцитов не происходит |
Формула : Т В+ NK+. Нарушены все звенья адаптивного иммунитета |
|
Х-сцепленная агаммагло булине- мия Брутона |
Btk (Хq) |
Дефект тирозинкиназы Btk, участвующей в передаче активационного сигнала в В-клет- ках и их дифференци- ровке. Блок развития В-клеток на стадии про-BII |
Формула : Т В- NK+. Нарушен адаптивный гуморальный иммунитет |
|
Гипер-IgM- синдром |
TNFSF5 (Хq) |
Нарушена экспрессия СD154 (при основной форме) или его рецептора CD40, что приводит к нарушению взаимодействия между Т-клетками с одной стороны и макрофагами, дендритными и В-клетками — с другой |
Нарушение переключения С-генов иммуноглобулинов, аффинтет BCR не повышается, повреждена активность дендритных клеток, Т-хелперов и макрофагов |
Продолжение табл. 4.19
|
Название |
Ген (хромосома) |
Природа дефекта |
Иммунологические проявления |
|
Общая вариабельная иммунная недостаточность |
TNFSF20, BLIMP1, ICOS |
Гетерогенная группа иммунодефицитов. В их основе могут лежать дефекты В-кле- точных факторов BAFF (фактор выживаемости В-клеток) или Blimp-1 (фактор дифференци- ровки плазмоцитов), а также костимулиру- ющей молекулы ICOS. Нарушен гомеостаз популяции В-лимфоци- тов и дифференцировка антителообразующих клеток |
Нарушение преимущественно гуморального иммунного ответа |
|
Селективный IgA дефицит |
Не установлен |
Нарушение переключения С-генов иммуноглобулинов на Са. Снижено содержание IgA, ответственного за защиту слизистых оболочек |
Нарушение гуморальной защиты слизистых оболочек |
|
Гипер-IgE- синдром |
Не установлен |
Природа повышения уровня IgE не установлена |
Гнойное воспаление. Проявления аллергии |
|
Сидром Вискотта—Олдрича |
WAS (Xp) |
Дефект белка WASP, важного для взаимодействия сигнальных молекул и белков цитоскелета. Нарушена подвижность клеток и формирование контактов |
Функциональные повреждения, приводящие к нарушению фагоцитоза, презентации антигена, контактного цитолиза |
|
Атаксия-телеанги эктазия |
ATM (11q) |
Дефект киназы АТМ, ответственной за репарацию разрывов ДНК и запуск апоптоза клеток при невозможности такой репарации |
Накопление клеток с нерепарированными разрывами ДНК. Среди разнообразных дефектов — нарушение развития лимфоцитов, особенно Т-клеток и обусловленная этим иммунная недостаточность |
|
Синдром Ниймегена |
NBS1 (7) |
Дефект гена NBS1, ответственного за репарацию разрывов ДНК, с последствиями как при дефекте гена АТМ |
То же |
Окончание табл. 4.19
|
Название |
Ген (хромосома) |
Природа дефекта |
Иммунологические проявления |
|
Аутоиммунный лимфопролиферативный синдром |
TNFSFR6 (10q) |
Дефицит генов, кодирующих Fas-рецептор (чаще всего), Fas-лиганд или каспазы 8 и 10. В результате нарушается рецепторный апоптоз, в том числе развивающихся клеток иммунной системы |
Аутоиммунные процессы, гиперпролиферация лимфоцитов, гипериммуноглобу- линемия, накопление CD3+ CD4- CD8- Т-клеток |
|
Х-сцепленный лимфопролиферативный синдром |
SH2D1A (Х) |
Дефект гена SH2D1A, следствием которого является повышение вероятности инфицирования В-клеток вирусом Эпштейна-Барр, вызывающим нарушения функций этих клеток |
Инфекционный мононуклеоз, лимфопролиферация, дисиммуногло- булинемия, нарушение гуморального иммунного ответа |
|
IPEX-синдром |
FOXP3 (Хр) |
Дефект гена FOXP3, ответственного за развитие регуляторных Т-клеток, предотвращающих аутоиммунные процессы и любые избыточные иммунные процессы |
Развитие множественных аутоиммунных процессов (особенно повреждающих эндокринные органы), гиперпролиферация лимфоцитов кишечника |
|
APECED-синдром |
AIRE |
Дефект гена AIRE, ответственного за экспрессию в тимусе различных органоспецифических антигенов. Усиление поступления на периферию аутоспецифических Т-клеток |
Аутоиммунные эндо- кринопатии |
Различают 16 вариантов ТКИН.
• Х-сцепленная тяжелая комбинированная иммунная недостаточность [дефицит у(с)-цепи]. Вызвана мутацией локализующегося в Х-хромосоме гена у(с)-цепи, общей для рецепторов для IL-2, IL-4, IL-7, IL-9, IL-15 и IL-21 (рис. 4.43). Самый частый вариант ТКИН — 46% от всех ее случаев. При этой форме иммунодефицита теоретически не должны проявлять свою активность все перечисленные цитокины (из-за дефекта их рецепторов). Однако в реальности важно отсутствие функций только одного цитокина — IL-7, в связи с тем, что он обеспечивает выживаемость лимфоидных предшественников и развитие всех ростков лимфопоэза. У мышей, гомозиготных по мутации scid, отсутствуют Т-, В- и NK-клетки. У человека роль IL-7 в развитии В-лимфоцитов заменима, и больные с Х-сцепленной формой ТКИН не имеют Т- и NK-клеток, тогда как В-лимфоциты у них развиваются практически нормально. Развитие Т-клеток блокируется на стадии перехода DN1 в DN2.
- Дефицит тирозинкиназы Jak3. Киназа Jak3 отвечает за передачу сигнала от у(с)-цепи (см. рис. 4.43). Именно поэтому фенотипические проявления ее дефицита практически совпадают с таковыми для Х-сцепленной ТКИН с той же формулой развития лимфоцитарных ростков: Т В+ NK-.
- Дефицит CD45. CD45 — крупная молекула, функционально связанная с рецепторным комплексом, цитоплазматическая часть которой обладает активностью тирозинфосфатазы. Она выполняет важную роль при активации Т-лимфоцитов, катализируя дефосфорилирование молекулы тирозинкиназы Lck (в покоящихся клетках эта молекула функционально неактивна и находится в гиперфосфорилированном состоянии). Дефосфорилирование приводит к изменению конформации этой молекулы и переходу ее в функционально активное состояние (см. раздел 3.4.2.1). Если учесть ключевую роль тирозинкиназы Lck в запуске внутриклеточной передачи сигнала в Т-клетках, значение фосфатазы CD45 в пусковых механизмах активации становится понятным. Молекула CD45 выполняет и другие функции, в частности, при активации Т-клеток памяти, о чем свидетельствует изменение структуры внеклеточной части молекулы при дифференцировке этих клеток (см. раздел 3.5.3.2).
- Дисгенезия тимуса (синдром Незелоф). Частичная или полная агенезия тимуса с гипоплазией, нарушением архитектуры лимфоидной ткани и отсутствием зародышевых центров. Комбинированный иммунодефицит с нормальным или повышенным уровнем одного или нескольких классов иммуноглобулинов при ослаблении индуцированного синтеза антител. На Т-клетках ослаблена экспрессия CD3 и CD4, но усилена экспрессия CD44.
- Дефицит RAG-1/RAG-2. Рекомбиназы 1 и 2 формируют комплекс, отвественный за запуск перестройки V-генов антигенраспознающих рецепторов (см. раздел 3.1.4.1). Мутации генов RAG1 и RAG2 полностью блокируют процесс перестройки V-генов. В связи с этим развитие Т- и В-лимфоцитов останавливается на стадиях, предшествующих этому событию: для Т-клеток это стадия DN2, для В-клеток — про-В. Развитие NK-клеток не нарушено.
- Дефицит ДНК-зависимой протеинкиназы (радиочувствительная ТКИН). ДНК-зависимая протеинкиназа составляет часть рекомбинационного комплекса — комплекса факторов, необходимых для перестройки V-генов. Эти факторы экспрессируются перед началом реаранжировки. ДНК-зависимая протеинкиназа участвует в ликвидации (разрешении) «шпилек», формирующихся после двунитевых разрывов, когда разорванные нити замыкаются на самих себя (см. раздел 3.1.4.1). Разрешение «шпилек» состоит в разрывах этих связей, после чего устанавливаются окончательные связи между разорванными участками гомологичных
нитей ДНК. Формирование двунитевых разрывов происходит как в процессе реаранжировки V-генов, так и при действии повреждающих факторов, например, ионизирующей радиации. Именно поэтому мутации гена ДНК-зависимой протеинкиназы не только нарушают процесс реаранжировки V-генов, но и препятствуют ликвидации последствий действия радиации и других факторов, повреждающих ДНК. Блокада развития Т- и В-клеток происходит на тех же этапах, как при дефиците RAG-1/RAG-2.
- Дефицит a-цепи рецептора IL-7. Механизм формирования иммунодефицита в принципе таков же, как при дефекте у(с)-цепи, хотя он касается только проявления действия IL-7 (см. рис. 4.43). В отсутствие сигналов, опосредованных этим цитокином, не происходит развития Т- и NK-клеток (у мышей — также В-клеток) в связи с апоптотической гибелью клеток- предшественников и отсутствием стимулов к пролиферации.
- Дефицит молекул MHC-II. Развивается в результате мутации генов трансактиватора генов класса II (CIITA — MHC class II transactivator, контролирует активность генов MHC класса II), а также генов регуляторных факторов семейства RFX — RFXNK, RFX5, RFXAP. Отсутствие экспрессии молекул MHC-II служит препятствием для дифференци- ровки и селекции субпопуляции CD4+ Т-клеток.
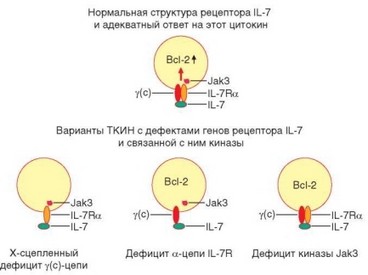
Рис. 4.43. Схема дефектов рецептора IL-7 и связанной с ним киназы при различных вариантах тяжелого комбинированного иммунодефицита. Выпадение любого из компонентов рецептора IL-7 или связанных с ним сигнальных факторов имеет идентичные последствия: вследствие отсутствия передачи сигнала экспрессии антиапоп- тотического фактора Bcl-2 не происходит, и клетки подвергаются апоптозу
- Дефицит ТАР. Молекулы ТАР обеспечивают транспорт пептидов, формирующихся в протеасомах, в эндоплазматический ретикулум, где они включаются в состав молекул MHC-I. Поскольку в отсутствие пептида молекулы MHC-I нестабильны, нарушается их экспрессия на поверхности клеток. Это служит причиной нарушения дифференцировки и селекции субпопуляции CD8+ Т-лимфоцитов.
- Дефицит CD25. Молекула CD25 — a-цепь рецептора IL-2, придающая ему высокое сродство к антигенам. В норме она экспрессируется на активированных Т-лимфоцитах (менее закономерно — на В-клетках), а также на неактивированных естественных регуляторных Т-клетках. Отсутствие этой молекулы затрудняет реализацию IL-2-зависимых процессов — экспансию клонов активированных Т-лимфоцитов и развитие естественных регуляторных Т-клеток. Численность лимфоцитов всех типов не изменена.
- Дефицит CD3y. Единственная форма генетических дефектов, затрагивающих комплекс CD3. Отсутствие у-цепи нарушает экспрессию комплекса TCR—CD3, для которой требуется полноценный субъединичный состав. Поскольку у-цепь, наряду с другими полипептидными цепями CD3, участвует в формировании активационного сигнала непосредственно после связывания антигенного лиганда с рецептором TCR, ее дефицит проявляется в ослаблении реактивности Т-клеток в ответ на антигенную стимуляцию.
- Дефицит ZAP-70. Тирозинкиназа ZAP-70 (семейство Syk) — один из ключевых факторов передачи активационных сигналов в Т-клетках (см. раздел 3.4.2.1). Она завершает первый этап передачи сигнала, являющийся общим для всех ветвей сигнальных путей, формирующихся дистальнее. Поэтому дефект гена ZAP-70 нарушает все проявления ответа Т-клеток. Кроме того, ZAP-70 участвует в передаче сигналов, необходимых для развития CD8+ Т-лимфоцитов, что обусловливает преимущественный дефицит этих лимфоцитов. Клеточная формула иммунодефицита: ^CD4+ CD8-) B+ NK+ .
- Дефицит CD4. Мутации гена корецептора CD4 препятствуют развитию субпопуляции Т-хелперов, распознающая функция которых зависит от CD4.
- Синдром Оменна (Omenn syndrome). Неполная блокада RAG вследствие мутаций генов RAG1 и RAG2, когда функция этих генов частично сохранена и происходит VDJ-рекомбинация, необходимая для создания антигенспецифического репертуара.
- Дефицит аденозиндезаминазы (ADA). Дефицит обусловлен мутацией гена ADA. Аденозиндезаминаза катализирует превращение аденозина и дезоксиаденозина соответственно в инозин и дезоксиинозин. В отсутствие аденозиндезаминазы накапливаются токсические метаболиты, из которых наиболее токсичен дезоксиаденозин. Накопление этих метаболитов вызывает повреждение Т- и В-лимфоцитов. Особенно чувствительны к аденозину и его метаболитам развивающиеся тимоци- ты, которые погибают под действием этих метаболитов. Дефицит аде- нозиндезаминазы был первым генетическим дефектом, устраненным путем генотерапии — переноса аутологичных клеток костного мозга, в которые был инкорпорирован ген ADA.
• Дефицит пуриннуклеотидфосфорилазы (PNP). Дефицит обусловлен мутацией гена PNP. Фермент пуриннуклеотидфосфорилаза катализирует превращение гуанозина в гуанин, а также инозина и дезоксиинозина в гипоксантин, что предотвращает накопление токсических продуктов метаболизма гуаниновых нуклеотидов — дезоксигуанозина и гуанозина, токсичных только для Т-лимфоцитов. Дефект пуриннуклеотидфос- форилазы частично компенсируется, в связи с чем он проявляется менее тяжело, чем дефицит аденозиндезаминазы.
Описанная группа ТКИН представляет наиболее тяжелые варианты первичных иммунодефицитов. В большинстве случаев больные дети погибают в течение первого года жизни от инфекций. Хотя не во всех случаях поражаются все линии лимфопоэза (неизменно поражение Т-ростка или субпопуляций Т-лимфоцитов) на уровне функционирования иммунной системы, иммунодефицит всегда является комбинированным в связи с зависимостью всех форм иммунного ответа от Т-лимфоцитов.