Синдром удлиненного QT (LQT)
Данное заболевание впервые в 1957 г. описали Jervell и Lange-Nielsen. Обследовав близких родственников, они пришли к заключению, что эта патология является наследственным заболеванием, которое характеризуется наличием удлиненного интервала QT на ЭКГ, врожденной глухоты и случаями возникновения внезапной смерти в детском возрасте. Спустя несколько лет Romano-Word сообщил о наличии другого варианта (аутосомно-доминантного) наследования этой патологии. У таких больных наблюдаются обмороки, случаи неожиданной (внезапной) смерти, пароксизмы «пируэт» формы желудочковой тахикардии, которая нередко провоцируется физическим или эмоциональным стрессом.
Современные молекулярные методы изучения наследственности выявили у таких больных наличие мутации генов, кодирующих функцию протеинов ионных каналов в миокарде. В настоящее время установлено более 100 различных мутаций в 5 генах, контролирующих функции ионных каналов сердца. Предполагается, что в ещё не изученных семьях, могут быть выявлены другие, сегодня неизвестные, виды нарушений.
Большинство из указанных мутаций гены контролируют функцию калиевых каналов. И только один ген SCN 5А ответственен за функцию натриевых каналов. Четыре гена из пяти в настоящее время установлены (табл. 9).
Синдром Jervel и Lange-Nielsen (JLNS) также генетически гетерогенен. Глухота передается по наследству по аутосомально-рецессивному типу, а удлинение QT по доминантному. Данный синдром возникает в результате мутаций в любой из двух взаимодействующих молекул (min К и Kv LQT 1).
Полученные данные о роли некоторых генов в регуляции транспорта ионов через клеточную мембрану позволяют высказывать предположение об их
Таблица 9
Генетическая характеристика различных типов удлиненного интервала QT (LQT)
|
Тип LQT |
Изменения в хромосоме |
Ответственный ген |
Ионные каналы |
|
LQT1 |
11 р15-5 |
KyLQT 1 |
Ю |
|
LQT2 |
7 g 35 |
HERG |
К+ |
|
LQT3 |
3 р 21 |
SCN 5А |
Na+ |
|
LQT4 |
7 g 25-27 |
? |
? |
|
LQT5 |
21 g 22 |
min К |
К+ |
|
JLNS 1 |
11 р 155 |
Kv LQT 1 |
К+ |
|
JLNS2 |
21 g 22 |
min К |
к+ |
влиянии на возникновение аритмогенного действия ряда лекарственных препаратов. В частности, возможно у таких больных имеются нарушения, выражающиеся в наличии повышенной чувствительности к действию препарата на некоторые калиевые каналы (Ikr или Iks). Наличие такой скрытой возможности демонстрируется данными, полученными при обследовании родителей больных с синдромом JLN. Этот синдром у ребенка возникает лишь в том случае, если ребенок наследует патологически измененный аллель от отца и матери, у которых нет клинических проявлений данной патологии.
Механизм аритмии. Удлинение QT у больных с данной патологией обусловлено удлинением клеточного потенциала действия. Но только удлинение интервала QT самостоятельно не может вызвать аритмию. Для этого необходимо также наличие выраженной дисперсии в реполяризации раз- личныхучастков миокарда и соответственно в их рефрактерности. При этом условии удлинение интервала QT может приводить к возникновению ранней постдеполяризации, т.е. к появлению желудочковой экстрасистолы, которая по механизму ри-энтри провоцирует развитие желудочковой тахикардии типа «пируэт».
Экспериментальные данные, пока не подтвержденные in vivo, позволяют предполагать, что ведущую роль в развитии удлиненного QT играют М клетки, располагающиеся в глубоких слоях миокарда. В этих клетках у млекопитающих было обнаружено, что по сравнению с клетками из эндокардиального слоя, потенциал действия в норме более продолжительный и эти клетки более чувствительны к препаратам удлиняющим реполяризацию. Поэтому высказывается мнение, что в тех случаях, когда под влиянием лекарственных средств увеличивается трансмуральная дисперсия в продолжительности реполяризации, появляются условия для возникновения реципрокной тахикардии.
В эксперименте на животных также было показано, что ишемия миокарда в сочетании с симатической стимуляцией также увеличивает дисперсию продолжительности рефрактерного периода.
У человека при использовании методики позволяющей проводить регистрацию внеклеточной электрограммы в различных участках миокарда, во время операции на открытом сердце было продемонстрировано также наличие нарастания дисперсии продолжительности рефрактерного периода у больных, у которых возникали кратковременные эпизоды фибрилляции предсердий или мономорфной желудочковой тахикардии, быстро трансформировавшейся в фибрилляцию желудочков.
В последние годы обсуждается роль электрического ремоделирования и кардиальной «памяти» в возникновении состояния, при котором более легко возникают и становятся более продолжительными повторные пароксиз
мы аритмии. Этот феномен связывают с синтезом нового белка в клетках в результате значительного увеличения активности циклогексимида и снижением ионного тока (Ito), что сопровождается удлинением потенциала действия и увеличением трансмиокардиальной дисперсии времени реполяризации клеток. Кроме того, изменения растяжения миокарда во время тахикардии активирует синтез в кардиальных клетках ангиотензина II, ренина, АПФ, которые стимулируют возникновение кардиальной «памяти», элект- рофизиологического ремоделирования.
Многие химические соединения, включая и лекарственные препараты, могут оказывать проаритмическое действие. Механизм такого действия заключается в возникновении блокады калиевых каналов (1кг), что вызывает уширение потенциала действия, удлинение интервала QT и появления ранней постдеполяризации и ри-энтри. Резко увеличивается вероятность данного нежелательного действия при комбинированной терапии препаратами, которые ингибируют цитохром Р-450 (СУР ЗА4), который участвует в метаболизме многих лекарственных средств. К таким препаратам относятся следующие группы препаратов:
- имидазолсодержащие противогрибковые средства;
- антибиотики (макролиды, флорквиналоны);
- антималярийные;
- трициклические антидепрессанты;
- нейролептики;
- антигистаминные (астемизол, терфенадин).
Предрасполагают к развитию удлинения QT и проаритмических эффектов следующие заболевания или состояния:
- врожденный синдром удлиненного QT;
- органические заболевания сердца, особенно при наличии сердечной недостаточности;
- гипокалиемия, гипомагнеземия;
- брадикардия;
- женский пол
Диагноз. Выделяют две группы критериев: большие и малые.
Большие включают следующие данные:
- удлинение (более 440 мс) коррегированного QT (рис. 29А);
- наличие в семье больных, имеющих удлинение интервала QT;
- обморок, вызванный стрессом;
- врожденная глухота в сочетании с удлинением интервала QT.
Малые критерии:
- врожденная глухота;
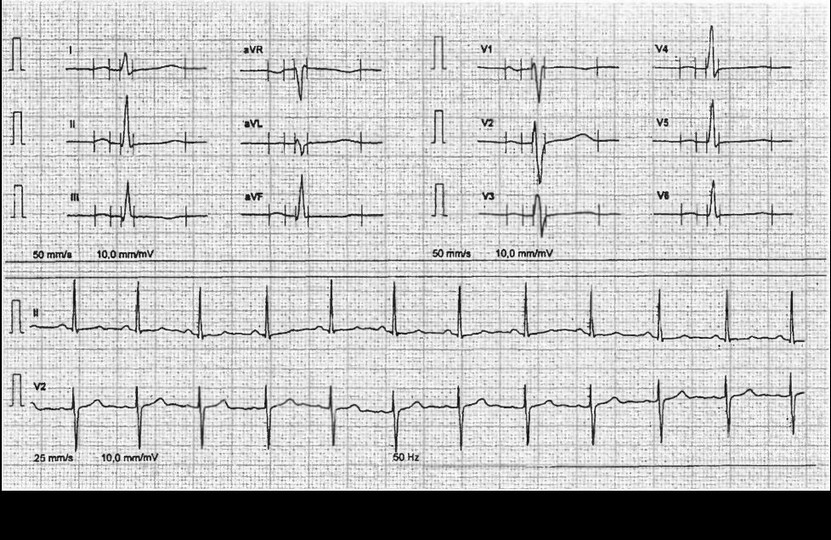
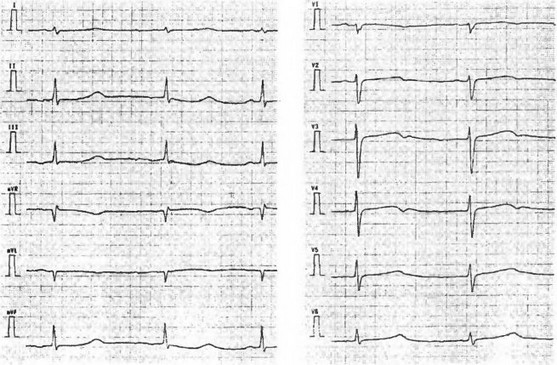
Рис. 29 Б. Синдром удлиненного QT.
Ритм синусовый с частотой 55 в 1 мин, интервал QT - 540 мс. Зубег Т отрицательный или двуфазный в отведениях a VL, V2-V4
- периодически возникающие изменения зубца Т (рис. 29Б);
- периодически возникающие изменения сегмента ST;
- брадикардия (в детском возрасте).
Клинические данные. Семейные случаи заболевания регистрируются примерно в 50% случаев. Наиболее часто встречается синдром Романо- Ворд'а (1:10-15 тыс. чел.); синдром Джервелл-Нильсен'а является очень редко встречающейся наследственной формой болезни.
Данные, полученные при обследовании относительно небольших групп больных с синдромом Romano-Word, позволили установить наличие связи между клиническими проявлениями и типом генетических нарушений (табл. 10).
Представленные в таблице три типа синдрома удлиненного QT (LQT) характеризуются аутосомно-доминантным типом наследования. Наиболее часто встречающимися являются первые 2 типа, при которых нарушается транспорт ионов калия (Ikr, Iks) через мембрану клеток.
Наиболее частым проявлением синдрома удлиненного QT являются обмороки. У многих больных они начинают возникать в детском возрасте (5-15 лет). В среднем обмороки раньше появляются у лиц мужского пола, чем
Таблица 10
Наиболее часто встречающиеся варианты генетических нарушений и особенности клинических проявлений
|
Клинические данные |
Тип 1 (LQT1) |
Тип 2 (LQT 2) |
Тип 3 (LQT 30) |
|
Мутация гена |
К LQT 1 |
HERG |
SCN SA |
|
Нарушения транспорта ионов |
Снижение Iks |
Снижение Ikr |
УвеличениеINa |
|
Возникновение симптомов (средний возраст) |
9 лет |
12 лет |
16 лет |
|
Факторы, провоцирующие возникновение обмороков |
Физический, эмоциональный |
Резкий звук, стресс |
В покое, во время сна |
|
Характер изменений зубца Т |
Умеренный |
Расщепленный |
Малый и заостренный |
|
Изменения интервала QT на нагрузке |
Не изменяется |
В нормальных пределах укорачивается |
Резко укорачивается |
у женщин. Их возникновение в возрасте менее 5 лет свидетельствует о более тяжелом течении заболевания. Особенно плохой прогноз наблюдается у тех, у которых обмороки стали возникать в течение первого года жизни. Пароксизм желудочковой аритмии, потребовавшей проведения реанимационных мероприятий (в частности, электрошока), увеличивает вероятность возникновения повторной остановки сердечной деятельности в ближайшее время более чем в 10 раз (по сравнению с теми, у кого остановка сердца не наблюдалась).
Семейный анамнез. Отсутствие случаев внезапной смерти у родственников в молодом возрасте не исключает вероятности её возникновения у данного больного. И наоборот, наличие таких случаев не свидетельствует об абсолютной фатальности течения такого заболевания. Поэтому предсказательное значение семейного анамнеза относительно невелико.
Отдаленные наблюдения за исходами у таких больных показали, что чем больше длительность интервала QT, тем выше риск внезапной смерти. Особенно риск увеличивается у тех больных, у которых продолжительность QT достигает более 600 мс. Однако наличие нормального интервала QT (менее 440 мс) у члена семьи, среди которых имеются больные с синдромом удлиненного QT, не исключает риска внезапной смерти или появления обмороков, свидетельствующих о появлении жизнеопасных нарушений желудочкового ритма. Величина такого риска относительно невелика (примерно 5%).
У мужчин фатальные нарушения развиваются раньше, чем у женщин. Если у мужчины к 20 годам не появляются обмороки, нарушения ритма сердца,
Таблица II
Стратификация риска внезапной смерти у больных с синдромам удлиненного QT
|
Класс доказательств |
Факторы риска |
|
Класс 1 (данные не вызывающие сомнений) |
|
|
Класс IIА (данные противоречивы, но доказательства пользы для прогнозирования преобладают) |
и атриовентрикулярной блокады |
|
Класс II Б (данные противоречивы, но больше данных, указывающих на отсутствие влияния на прогноз) |
|
то риск их возникновения в последующие годы жизни невелик. У женщин с возрастом величина риска не изменяется. Провоцирующим фактором риска внезапной смерти у женщин являются роды. В послеродовом периоде женщины из такой семьи даже в случае нормальной ЭКГ в течение 1 года должны принимать бета-адреноблокаторы.
На прогноз жизни у таких больных оказывают влияние многие факторы, включая характер генетических нарушений (табл. 11). При наличии синдрома Романо-Ворд'а прогноз значительно благоприятнее, чем у больных с синдромом Джервелла и Ланге-Нильсон'а. Кроме того, среди больных, имеющих синдром Романо-Ворд'а, прогноз также в значительной степени зависит от типа синдрома. Третий тип, при котором нарушается транспорт ионов натрия, протекает наиболее неблагоприятно и плохо поддается контролю с помощью бета-адреноблокаторов. При первом и втором типах синдрома наблюдается большая частота обмороков, однако летальность ниже. Эффективность терапии бета-адреноблокаторами достаточно высокая, особенно при первом типе.
Летальность среди больных, которые не получают адекватную терапию составляет не менее 20% в течение года после первого случая обморочного состояния и около 50% в течение 10 лет. Современная тактика ведения таких больных снижает риск внезапной смерти почти в 10 раз.
Профилактика желудочковой тахикардии и внезапной смерти. Все
больные и их родственники должны избегать занятий спортом, тяжелых физических нагрузок, воздействия неожиданных звуковых раздражителей, погружения в холодную воду. Противопоказан прием препаратов, которые могут удлинять интервал QT.
В настоящее время, несмотря на отсутствие проспективных исследований, нет дискуссии о пользе профилактической терапии с помощью бета- адреноблокаторов больных с удлиненным QT. Это связано с тем, что их эффективность очевидна и демонстрируется уменьшением примерно на 75% приступов обморочных состояний и уменьшением случаев внезапной смерти. Например, ретроспективный анализ данных, полученных в одном из самых больших исследований (233 больных), показал, что летальность в течение 15 лет в группе с обмороками в анамнезе составила лишь 9% на фоне терапии бета-адреноблокатором или после удаления симпатических ганглиев в нижнем шейном и в верхнем грудном отделах позвоночника слева, но достигла почти 60% среди тех, кто не получал указанного лечения.
Симпатическую денервацию рекомендуют проводить у тех больных, у которых, несмотря на прием блокаторов бета-адренорецепторов в высокой дозе, продолжают возникать обмороки, при наличии противопоказаний для их применения или при не согласии больного принимать эти препараты.
Наличие у больного сочетания синдрома удлиненного QT и нарушения атриовентрикулярной проводимости или брадикардии служит показанием для имплантации водителя ритма с последующим проведением лечения с помощью блокаторов бета-адренорецепторов.
В настоящее время нет отдаленных наблюдений с целью оценки эффективности имплантируемых кардиовертеров-дефибрилляторов у данной группы больных. Но, опираясь на результаты, полученные у других групп больных с высоким риском внезапной смерти, этот метод её профилактики рекомендуется использовать у больных, которые перенесли успешную реанимацию.
Обсуждается вопрос об имплантации кардиовертера-дефибриллятора у больных с синдромом Джервелл'а и Ланге-Нильсен'а и с полной поперечной блокадой.
При бессимптомном течении синдрома удлиненного QT, но при наличии прогностически неблагоприятных факторов, терапия бета-адреноблокато- рами также считается показанной. И даже при отсутствии удлинения QT оправданно использование этих препаратов в течение 1 года у женщины после родов, родственники которой имеют данное заболевание.
Идентификация патологических генов и связанных с ними нарушений транспорта ионов позволяют начать проведение исследований, направлен
ных на поиск подходов в лечении больных с различными генетическими типами данной патологии. Ретроспективный анализ данных указывает на то, что блокаторы бета-адренорецепторов вероятно обеспечивают наибольшее защитное действие при первом типе синдрома Романо-Ворд'а. Исходя из экспериментальных данных и первых клинических наблюдений, показавших, что лидокаин и мексилетин укорачивают интервал QT и нормализуют форму зубца Ту больных с третьим типом синдрома (нарушен транспорт натрия), необходимо проведение исследований с целью определения эффективности и безопасности этого класса препаратов при указанном генетическом типе патологии.
У больных со вторым типом синдрома Романо-Ворд'а (патология в хромосоме 7 HERG) отмечена нормализация ЭКГ данных (укорочение QT и нормализация зубца Т) при повышении содержания в крови ионов калия.
Источник: Мазур Н.А., «ПАРОКСИЗМАЛЬНЫЕ ТАХИКАРДИИ» 2005
А так же в разделе «Синдром удлиненного QT (LQT) »
- Предисловие
- Механизм возникновения аритмий
- Общие клинические методы обследования больных с подозрением на наличие пароксизмальной тахикардии
- Пароксизмальные наджелудочковые тахикардии
- Синусовая спонтанная тахикардия
- Синусоваяузловая реципрокная пароксизмальная тахикардия
- Фокальная (эктопическая) предсердная тахикардия
- Предсердная макро-ри-энтри тахикардия
- Предсердно-желудочковая узловая тахикардия
- Эктопическая (фокальная) тахикардия из атриовентрикулярного соединения
- Непароксизмальная реципрокная тахикардия из атриовентрикулярного соединения
- Пароксизмальная предсердно-желудочковая реципрокная тахикардия
- Фибрилляция и трепетание предсердий
- Желудочковые тахикардии
- Желудочковая тахикардия у больных без органического поражения сердца
- Синдром укороченного QT
- Аритмогенная правожелудочковая кардиомиопатия
- Синдром Бругада
- Трепетание и фибрилляция желудочков
- Внезапная смерть и ее профилактика
- Факторы риска возникновения фибрилляции желудочков у больных ИБС
- Профилактика ВСС у больных ИБС
- Гипертрофическая кардиомиопатия и риск ВСС
- Дилатационная кардиомиопатия, сердечная недостаточность и риск ВСС
- Клапанные пороки и риск внезапной смерти
- Аномалии отхождения коронарных артерий, мышечные мостики и риск внезапной смерти
- Нарушение функции проводящей системы сердца
- Внезапная смерть после аблации предсердно-желудочкового узла и после имплантации водителя ритма
- ВСС у людей со «здоровым» сердцем
- Реанимация в амбулаторных условиях
- Антиаритмические препараты
- Выбор антиаритмической терапии, её безопасность и эффективность при длительном лечении
- Литература
- Приложение Алгоритмы дифференциальной диагностики и выбора терапии